在配备UV和QDa检测器的ACQUITY Arc上使用系统性筛选策略对10种抗生素化合物进行方法开发
摘要
开发全新的LC和LC-MS方法即使对于经验丰富的分析人员同样具有挑战性,如果缺少专用策略,分析人员可能不知道从何入手或采取哪些步骤来开发新方法。常见方法开发策略包括筛选多种固定相、不同的流动相添加剂、不同的pH值、甚至不同的强溶剂。方法开发的复杂性因方法开发所采用的策略不同可能会加剧,不同分析人员可能采用不同的策略处理任务,由此增加不确定性并可能导致不同的最终结果。
本文所述研究重点介绍基于分层筛选策略的方法开发策略的优势及其在开发10种抗生素化合物分离方法中的应用。由于被测化合物的性质不同,某些化合物可能缺少UV发色团,因此需要同时采用UV和MS检测。利用同时配备PDA检测器和ACQUITY QDa质谱检测器的ACQUITY Arc系统开发方法。分层策略首先测试并评估流动相pH以确定性能更出色的条件。锁定流动相pH后,使用与确定的流动相兼容的色谱柱进行更传统的色谱柱筛选实验。在色谱柱筛选过程中测试乙腈和甲醇流动相,确定合适的分离条件。只需不到两个工作日即可开发出同时适用于LC-UV和LC-MS分析的最终方法,凸显了专用方法开发策略的速度优势。
优势
- 使用分层筛选策略快速执行方法开发活动
- 使用ACQUITY QDa在方法开发过程中追踪色谱峰,无需运行单标
- 使用ACQUITY QDa作为正交检测技术检测不含UV发色团的化合物
- 10种通用抗生素化合物均实现基线分离
简介
策略性方法开发过程可显著提升方法开发活动的速度和效率。常用方法开发策略可以分为四类:每次考察一种因素(OFAT)、全因子筛选(FFS)、分层筛选(TS)和分析方法质量源于设计(AQbD)。这些策略各有其优缺点,例如需要经验丰富的分析人员或使用不同类型的软件。OFAT需要由经验丰富的分析人员来解析结果并确定要试验的其他条件。如果样品特别复杂,或者分析人员不熟悉方法开发技术,这一策略可能相当耗时。FFS是一种全面策略,会测试固定相、流动相pH和流动相强溶剂的所有组合1-2。 由于需要探索各方面分离条件,因此数据采集和数据解析可能非常耗时。AQbD策略近期引起了广泛关注,但需要采用特殊软件,例如Fusion或DryLab3-5。 这些软件虽然需要一些培训,但最终可以得到一种稳定、完善的方法。AQbD使用高度依赖统计学的方法开发策略,可生成高度稳定的数据。在开发过程中考察方法稳定性,确保细微变化不会导致截然不同的结果。稳定的方法有助于减少返工、重新验证工作和方法失败率。此外,如果稳定性已通过测试并被证明足以满足要求,则可以更快速地进行方法验证。最后,TS策略在OFAT的速度与FFS的全面测试之间取得了平衡6。 分层筛选的方法开发活动逐步实施,每次实验筛选之后都包含一个决策步骤。每次测试一种变量,然后“锁定”该变量以对其他变量进行后续测试。
为展示分层筛选策略的应用,制备了由10种抗生素化合物组成的混合物,分别为阿莫西林、克拉维酸、哌拉西林、他唑巴坦、氨苄西林、苯唑西林、氯唑西林、双氯西林、阿奇霉素和头孢氨苄。这些化合物中有一些美国常用的处方用抗生素7,以及未来几年内专利即将到期的两种化合物(哌拉西林和他唑巴坦)。为分离这些化合物,测试了四种不同的固定相,以及两种强溶剂和两种流动相pH添加剂。在测试的第二天根据分层筛选策略选择最终方法,无需进一步优化分离条件。
实验
样品描述
10种抗生素购自Sigma Aldrich,用90:10水:乙腈稀释至以下浓度:阿莫西林、头孢氨苄、氨苄西林、苯唑西林、氯唑西林、双氯西林、哌拉西林和他唑巴坦95 µg/mL,阿奇霉素40 µg/mL,克拉维酸200 µg/mL。
方法条件
液相色谱条件
|
液相色谱系统: |
配备2998 PDA检测器和ACQUITY QDa质谱检测器的ACQUITY Arc |
|
检测条件: |
UV,215 nm,吸光度-MBF 抗生素化合物的SIR |
|
样品瓶: |
LCMS认证透明玻璃样品瓶,容积2 mL(部件号:600000751CV) |
|
色谱柱: |
XSelect CSH C18色谱柱, 3.0 × 50 mm, 2.5 µm(部件号:186006105) XSelect CSH苯己基柱, 3.0 × 50 mm, 2.5 µm(部件号:186006129) XSelect CSH氟苯基柱, 3.0 × 50 mm, 2.5 µm(部件号:186006117) XSelect HSS C18 SB色谱柱, 3.0 × 50 mm, 2.5 µm(部件号:186006165) |
|
柱温: |
30 °C |
|
样品温度: |
10 °C |
|
进样体积: |
3 µL |
|
流速: |
0.85 mL/min |
|
流动相A: |
0.1%甲酸(pH约3)或10 mM氢氧化铵(pH约10)的水溶液 |
|
流动相B: |
0.1%甲酸或10 mM氢氧化铵的乙腈溶液 |
|
流动相C: |
0.1%甲酸或10 mM氢氧化铵的甲醇溶液 |
梯度表
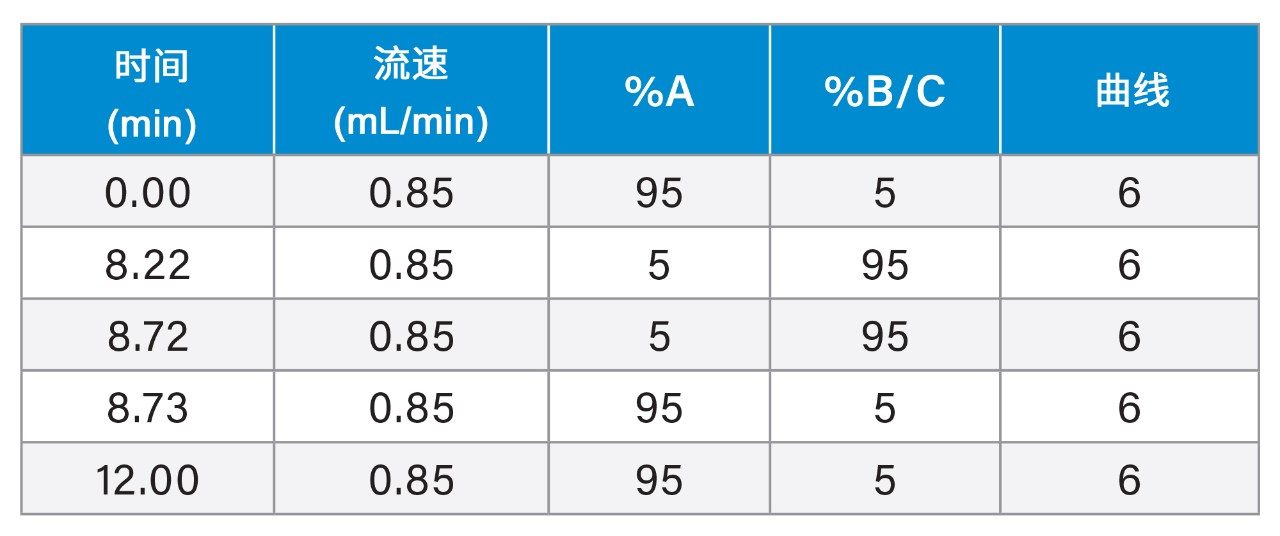
质谱条件
|
质谱系统: |
ACQUITY QDa质谱检测器 |
|
电离模式: |
SIR,ESI+模式 |
|
采集范围: |
175 m/z–550 m/z(采集的SIR如图1所示) |
|
毛细管电压: |
1.5 kV |
|
锥孔电压: |
15 V |
数据管理
|
色谱软件: |
Empower 3 Feature Release 4 |
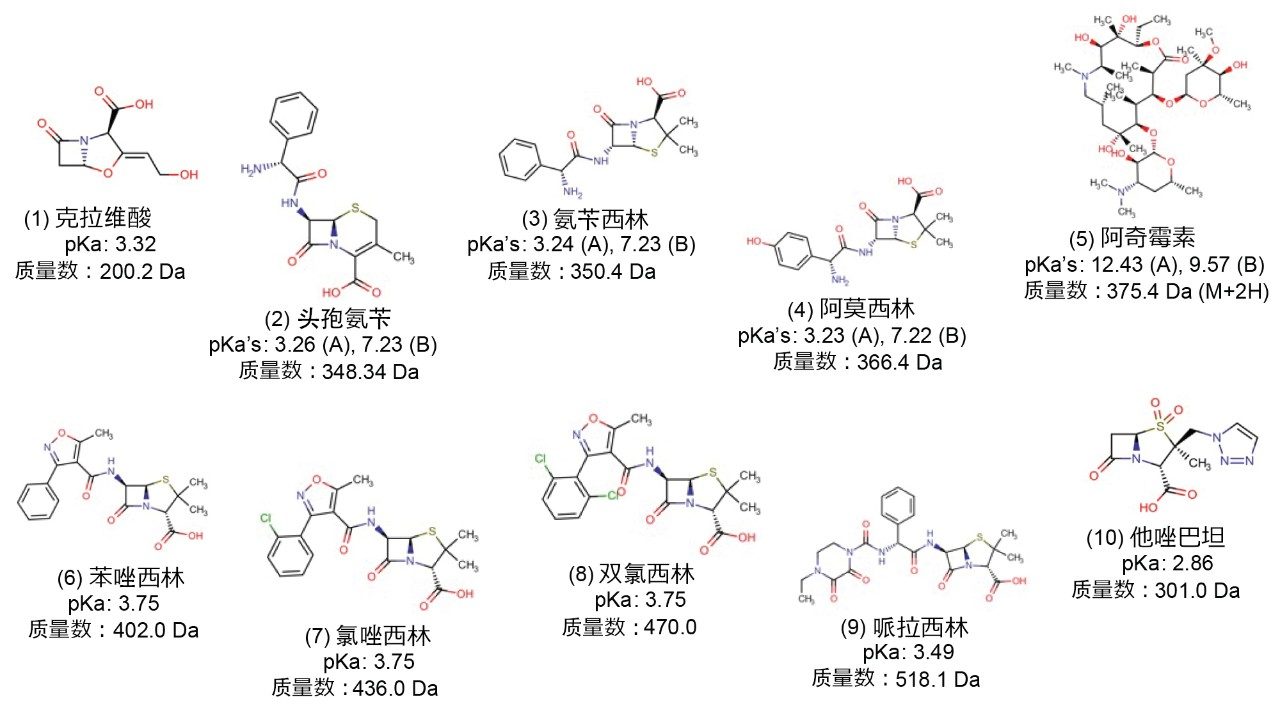 图1.样品混合物中10种抗生素的化学结构。所示pKa值由ChemAxon软件Chemicalize计算得出,并在适用情况下列为强酸(A)和强碱(B)。除非注明,否则所有未标记的pKa均为酸性pKa,列出的质量数均为用于通过SIR检测组分的荷电(M+H)质量数。
图1.样品混合物中10种抗生素的化学结构。所示pKa值由ChemAxon软件Chemicalize计算得出,并在适用情况下列为强酸(A)和强碱(B)。除非注明,否则所有未标记的pKa均为酸性pKa,列出的质量数均为用于通过SIR检测组分的荷电(M+H)质量数。
结果与讨论
分层筛选(TS)策略会执行不同“级别”或“层级”的方法开发实验。每个“层级”通常测试一种或两种参数,即流动相pH或固定相填料。完成每组实验之后,必须根据所获得的结果决定如何继续测试。所述研究中使用的分层筛选流程见图2。第一“层”测试流动相pH,并检查测试探针的保留结果。根据这些数据选择流动相pH进行后续测试。这种分步策略使分析人员可以测试分析适用的条件。
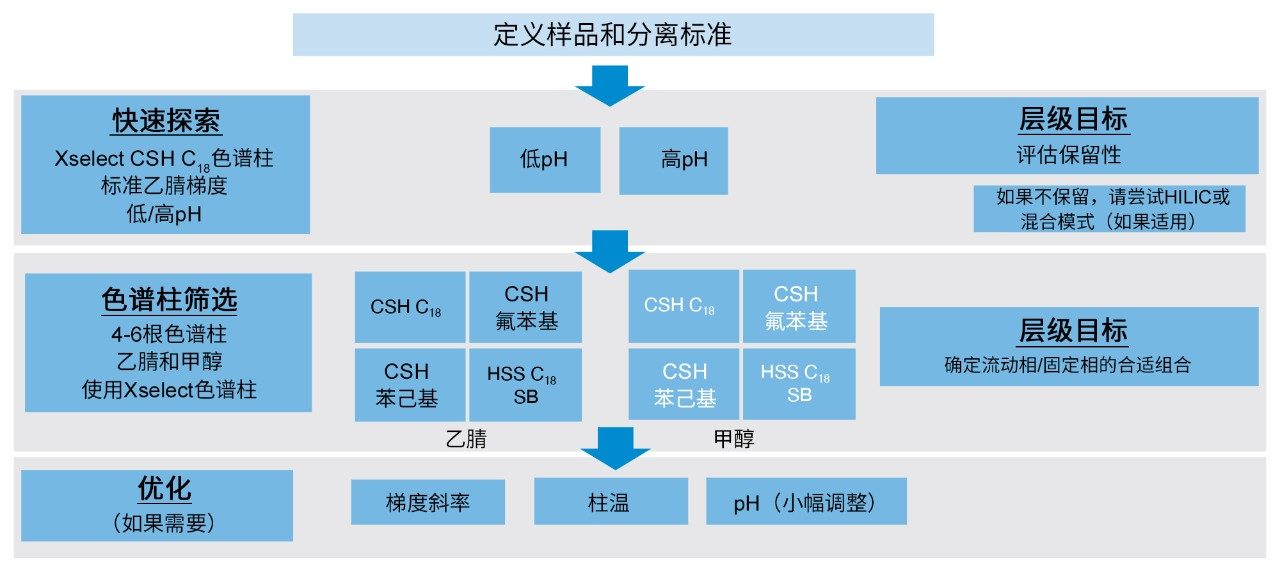 图2.开发分离10种抗生素化合物的方法使用的分层筛选策略
图2.开发分离10种抗生素化合物的方法使用的分层筛选策略
方法开发的第一步是定义样品和分离标准,由此在开发过程中设定一个明确的停止点。分离标准示例包括但不限于:相邻洗脱峰达到特定的USP分离度值或呈现良好的峰形。抗生素分离则必须达到以下三个目标才能获得可接受的方法。首先,所有化合物的USP分离度必须大于1.5(通常视为基线分离)。其次,化合物必须具有良好的峰形,峰对称性尽可能接近1。最后,化合物必须得到保留。
设定分离标准后,即可实施分层筛选的第一步。这一步通常称为快速探索,即使用低pH和高pH流动相添加剂来评估探针的保留性。在该步骤中,如果无论采用高pH还是低pH均无法实现充分保留,则可能需要采用替代色谱技术,例如混合模式色谱或亲水作用液相色谱(HILIC)。为执行快速探索,需要采用同时兼容高pH和低pH流动相的固定相。本例选择XSelect CSH C18色谱柱进行快速探索。该色谱柱填充有杂化有机硅胶固定相,能够在pH 1–11的范围内保持稳定,因此适用于探索实验。图3所示为使用XSelect CSH C18色谱柱和乙腈流动相针对10种抗生素进行快速探索实验得到的结果。
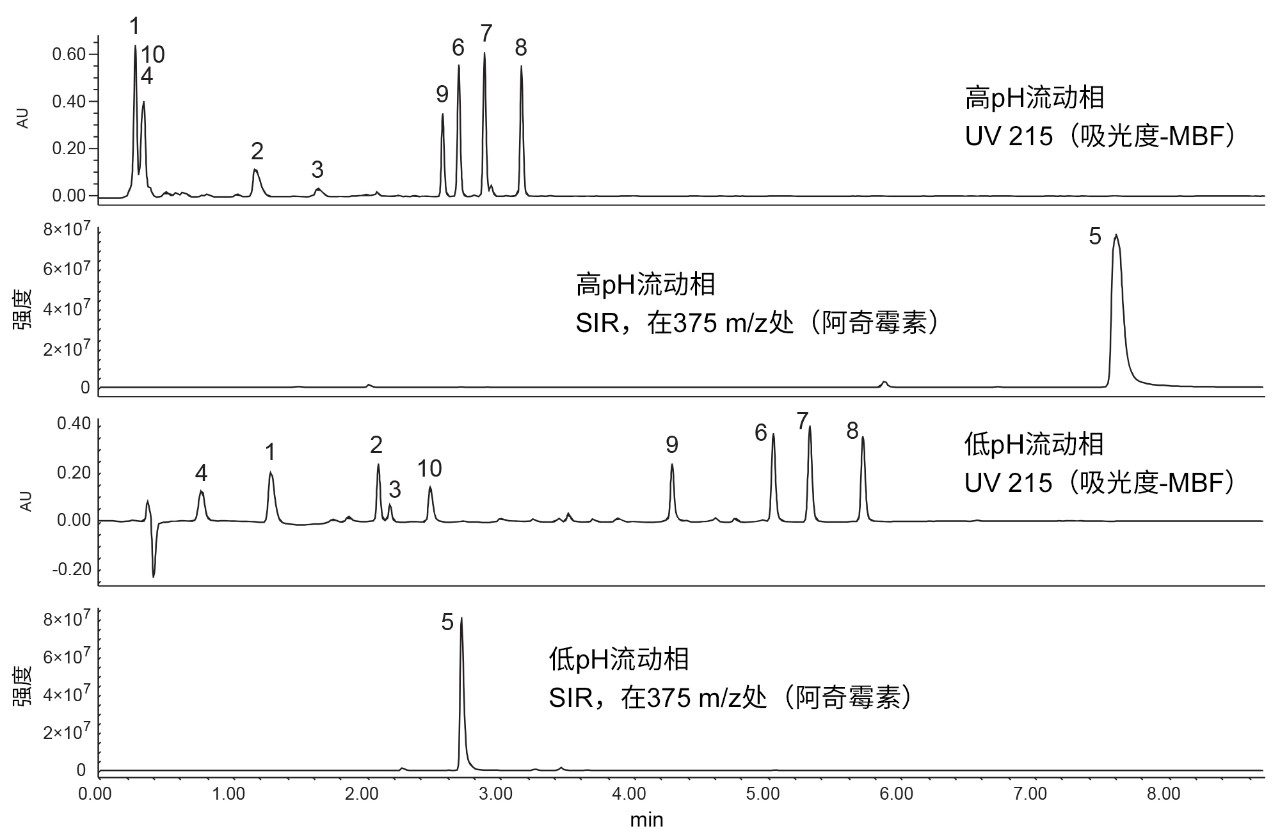 图3.使用XSelect CSH C18色谱柱与高pH(上图)和低pH(下图)流动相添加剂得到的10种抗生素的UV色谱图以及阿奇霉素(一种不含发色团的化合物)的SIR。1)克拉维酸,2)头孢氨苄,3)氨苄西林,4)阿莫西林,5)阿奇霉素,6)苯唑西林,7)氯唑西林,8)双氯西林,9)哌拉西林,10)他唑巴坦。
图3.使用XSelect CSH C18色谱柱与高pH(上图)和低pH(下图)流动相添加剂得到的10种抗生素的UV色谱图以及阿奇霉素(一种不含发色团的化合物)的SIR。1)克拉维酸,2)头孢氨苄,3)氨苄西林,4)阿莫西林,5)阿奇霉素,6)苯唑西林,7)氯唑西林,8)双氯西林,9)哌拉西林,10)他唑巴坦。
利用UV检测分析除阿奇霉素外的所有其他化合物;阿奇霉素是一种不含发色团的化合物,通过MS检测进行评估。利用吸光度-MBF降低215 nm波长下甲酸的背景噪音。从图3可以观察到高pH和低pH流动相添加剂之间的选择性差异。其中阿奇霉素(5)表现的差异尤为显著,低pH条件下该化合物在3 min之前洗脱,高pH条件下则几乎在8 min时洗脱。在低pH条件下,阿奇霉素的胺基带有双电荷;但在高pH条件下,胺基不再带电,化合物呈中性,因此具有更高的保留性。两种pH条件下阿奇霉素的电离效率也有所不同。在高pH条件下,阿奇霉素电离效率更高,但所得的峰较宽;而在低pH条件下,信号强度大致相同,但所得的峰较窄。他唑巴坦在低pH条件下部分带电且保留性良好,在约2.4 min时洗脱。在高pH条件下,他唑巴坦完全带电,因此极性更强,保留性下降。大多数化合物均观察到类似趋势,即在高pH条件下的保留性弱于低pH条件下的保留性。虽然这一趋势主要是由于化合物的电荷态,但也部分受到XSelect CSH C18色谱柱混合模式功能的影响。
XSelect CSH C18色谱柱使用含部分正电荷的杂化基质颗粒,这种修饰有助于改善碱性化合物在低pH条件下的峰形,并提供与酸性探针的混合模式相互作用。在低pH条件下,固定相的阴离子交换功能有助于保留酸性探针;而在高pH条件下,阴离子交换功能下降。苯唑西林(6)等化合物由于具有酸性或碱性官能团,因此在高pH条件下的保留性较低。10种化合物中有8种在低pH条件下得到完全分离,获得良好的保留和出色的峰形。因此,对抗生素分离选择低pH流动相条件。在其他情况下,高pH流动相可能更有利,例如阿奇霉素定量分析。
分层筛选的下一步是锁定选择的流动相pH,然后同时使用乙腈和甲醇流动相筛选一组合适的色谱柱。图4和图5分别为使用乙腈和甲醇流动相在选择的四根色谱柱上得到的抗生素分离结果,图中仅展示了215 nm UV通道,阿奇霉素的保留用组分编号(5)的位置来标记。
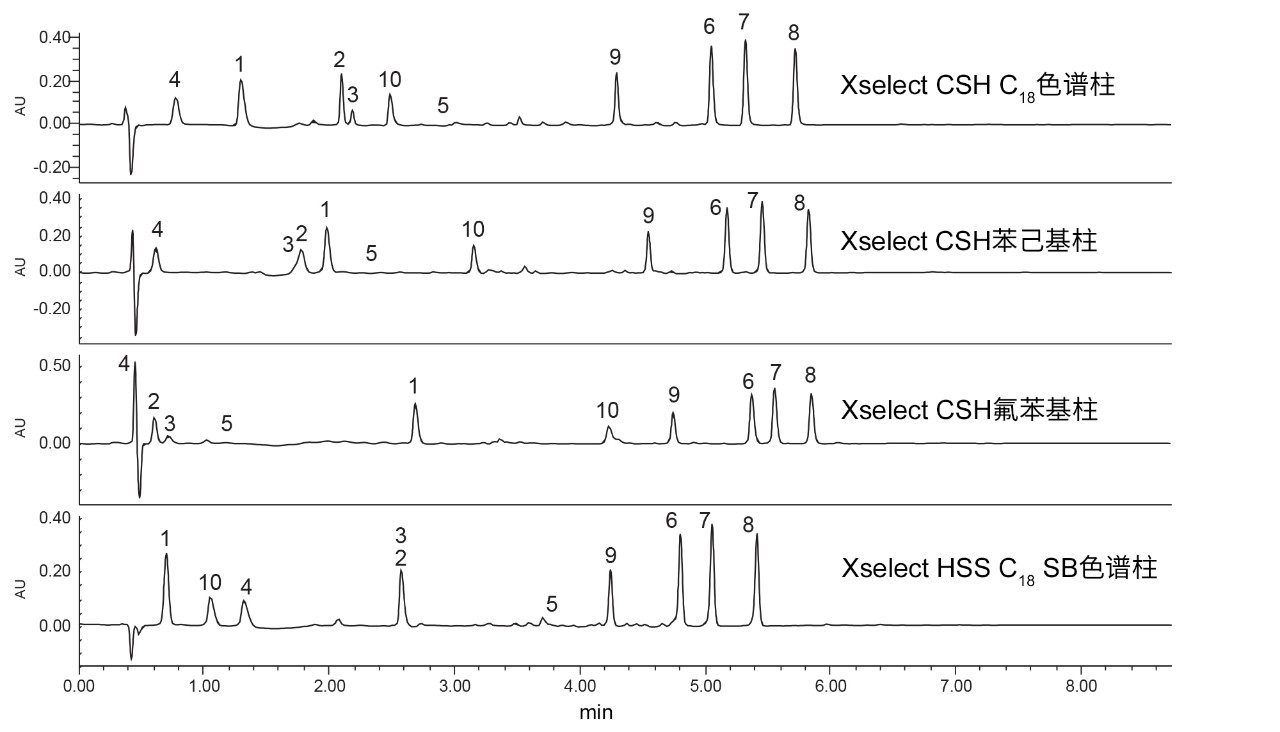 图4.使用乙腈作为强溶剂在四种固定相上得到的10种抗生素的分离结果。1)克拉维酸,2)头孢氨苄,3)氨苄西林,4)阿莫西林,5)阿奇霉素,6)苯唑西林,7)氯唑西林,8)双氯西林,9)哌拉西林,10)他唑巴坦。
图4.使用乙腈作为强溶剂在四种固定相上得到的10种抗生素的分离结果。1)克拉维酸,2)头孢氨苄,3)氨苄西林,4)阿莫西林,5)阿奇霉素,6)苯唑西林,7)氯唑西林,8)双氯西林,9)哌拉西林,10)他唑巴坦。
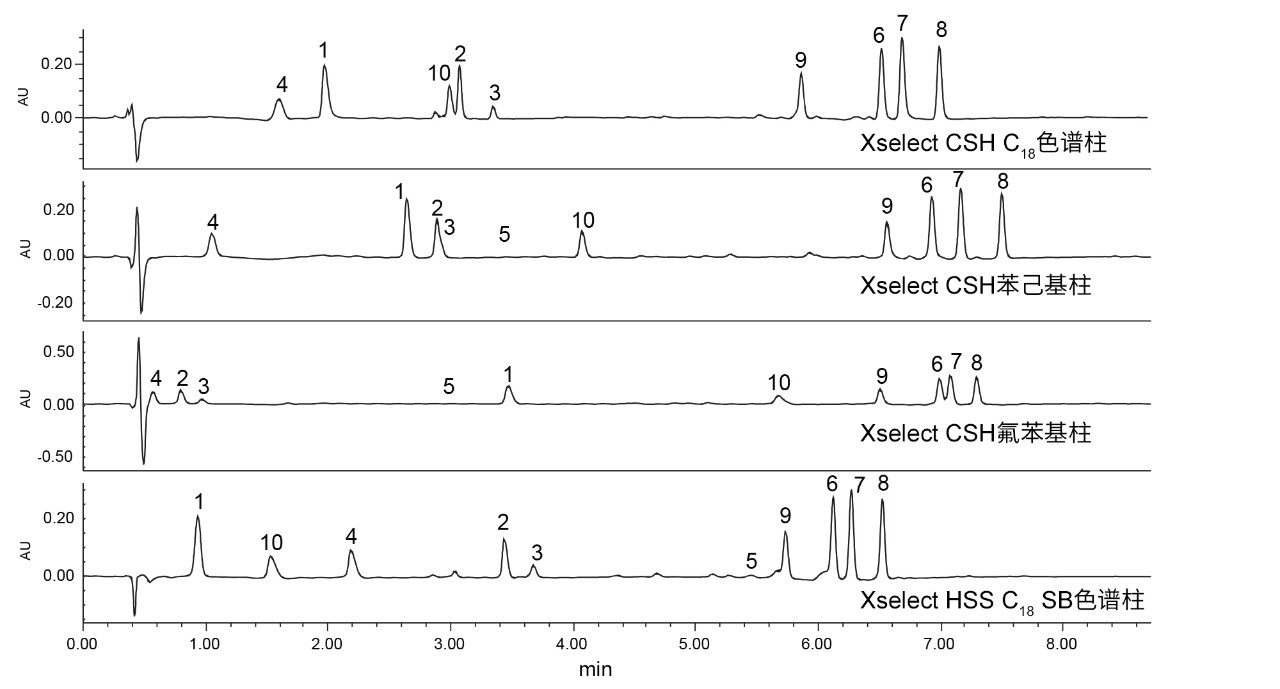 图5.使用甲醇作为强溶剂在四种固定相上得到的10种抗生素的分离结果。1)克拉维酸,2)头孢氨苄,3)氨苄西林,4)阿莫西林,5)阿奇霉素,6)苯唑西林,7)氯唑西林,8)双氯西林,9)哌拉西林,10)他唑巴坦。
图5.使用甲醇作为强溶剂在四种固定相上得到的10种抗生素的分离结果。1)克拉维酸,2)头孢氨苄,3)氨苄西林,4)阿莫西林,5)阿奇霉素,6)苯唑西林,7)氯唑西林,8)双氯西林,9)哌拉西林,10)他唑巴坦。
从这些图中可以看出,组分2和组分3(头孢氨苄和氨苄西林)难以分离。XSelect CSH氟苯基柱和XSelect CSH C18色谱柱都能够分离这两种化合物。氟苯基固定相对阿莫西林的保留性较差,且对头孢氨苄、氨苄西林和阿奇霉素的总体保留性较弱,这可能是因为低pH条件下这些探针具有碱性官能团,并且与基质颗粒上的正电荷发生相互作用。而在C18和苯己基固定相上的相互作用程度较弱。XSelect HSS C18 SB色谱柱为头孢氨苄、氨苄西林和阿莫西林提供了理想保留性能,凸显出基质颗粒在这些化合物的保留方面发挥的重要作用。只有在使用XSelect CSH C18色谱柱时,使用乙腈作为强溶剂才能满足方法开发开始时设定的标准。关键化合物对头孢氨苄和氨苄西林的最低USP分离度值为1.92,同时这些化合物也获得了良好的峰形和保留性能。XSelect CSH氟苯基固定相紧随其后,使所有化合物均实现良好分离(所有探针的USP分离度均>1.5),但保留性不佳。相同色谱柱使用甲醇流动相时产生的分离结果可能优于使用乙腈得到的结果。
甲醇流动相表现出与乙腈不同的结果。虽然预计甲醇会增强保留性,但不一定总能观察到选择性差异。不过,当使用苯己基固定相(例如XSelect CSH苯己基固定相)时,以甲醇作为强溶剂可提供一些特有的选择性差异。比较在XSelect CSH苯己基色谱柱上使用甲醇和乙腈得到的分离结果,发现洗脱顺序发生了变化。使用乙腈流动相时,克拉维酸(1)、头孢氨苄(2)和氨苄西林(3)按3、2、1的顺序洗脱。但是,使用甲醇时,洗脱顺序变为1、2、3。这种选择性变化是由于化合物与固定相之间发生次级相互作用,头孢氨苄和氨苄西林均含苯环,当使用甲醇时,苯环可通过π-π相互作用与苯己基固定相相互作用。
使用甲醇流动相时,XSelect CSH C18色谱柱和XSelect HSS C18 SB色谱柱都能够保留和分离所有化合物,而使用另外两种色谱柱时,观察到色谱峰共流出。XSelect CSH C18色谱柱使组分10和2(他唑巴坦和头孢氨苄)发生轻微的共流出,USP分离度为1.38,这一结果有可能在优化步骤中改善。XSelect HSS C18 SB色谱柱虽然能够保留和分离所有化合物,但组分9、6和10的峰形不佳。这些峰形在空白进样中不可见,很可能是由与固定相的相互作用引起的。充分考虑用乙腈获得的分离结果以及用甲醇获得的结果后选择一组最终检测条件。这些化合物在低pH条件下使用XSelect CSH C18色谱柱和乙腈流动相可获得理想分离结果,也可以使用相同色谱柱以甲醇作为强溶剂,但需要优化条件才能更好地分离关键化合物对。由于使用乙腈和XSelect CSH C18色谱柱已经满足所有设定标准,因此不需要对这一分离结果进行优化。图6展示了使用最终方法条件得到的10种化合物各自的SIR。
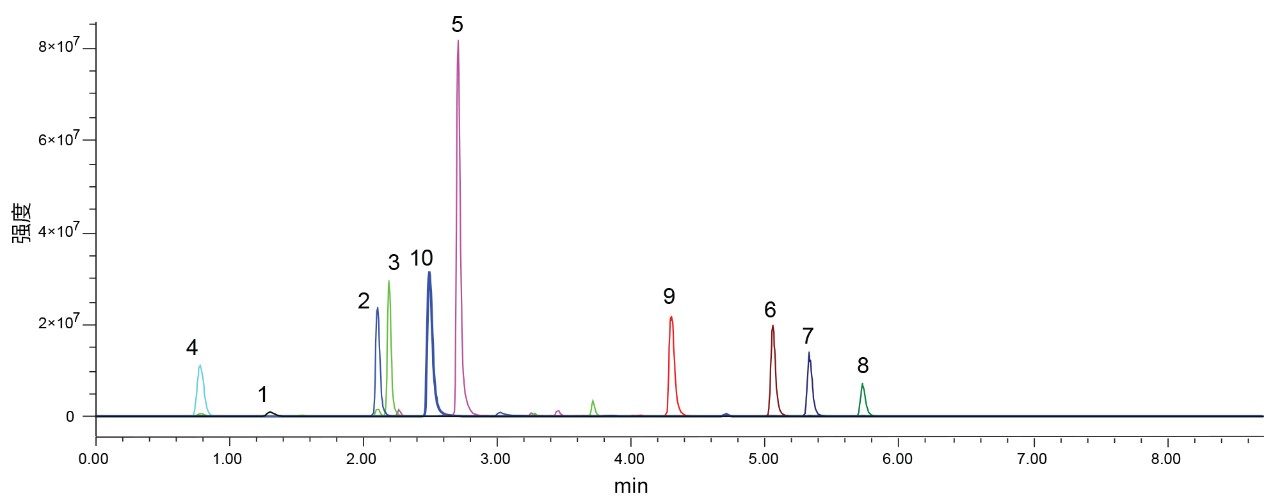 图6.在XSelect CSH C18色谱柱(3.0 × 50 mm, 2.5 µm)上分离10种抗生素化合物得到的10个SIR通道叠加图。含甲酸的水和乙腈流动相的流速为0.85 mL/min。梯度条件为:乙腈在8.22 min内从5%增加至95%。1)克拉维酸,2)头孢氨苄,3)氨苄西林,4)阿莫西林,5)阿奇霉素,6)苯唑西林,7)氯唑西林,8)双氯西林,9)哌拉西林,10)他唑巴坦。
图6.在XSelect CSH C18色谱柱(3.0 × 50 mm, 2.5 µm)上分离10种抗生素化合物得到的10个SIR通道叠加图。含甲酸的水和乙腈流动相的流速为0.85 mL/min。梯度条件为:乙腈在8.22 min内从5%增加至95%。1)克拉维酸,2)头孢氨苄,3)氨苄西林,4)阿莫西林,5)阿奇霉素,6)苯唑西林,7)氯唑西林,8)双氯西林,9)哌拉西林,10)他唑巴坦。
分层筛选方法开发策略在速度与样品全面检测之间取得了平衡。本研究在第一天实施快速探索,以便在高pH和低pH流动相之间切换时达到适当的色谱柱平衡。然后将色谱柱筛选实验设置为过夜运行并持续到第二天。最后,即使算上12 min运行时间,完成最终分离方法的开发也只用了不到两天的时间。
结论
方法开发使用的系统性筛选策略在整个过程中采用多“层”步骤,每“层”测试一组条件以确定能否提供更出色的结果。然后“锁定”这些条件用于后续测试。使用一种由10种常用处方抗生素组成的混合物,证明该方法开发策略的有效性。XSelect CSH C18色谱柱与乙腈流动相和低pH流动相添加剂甲酸使该混合物得到基线分离,获得了良好的保留和峰形。兼用UV和MS检测,可以在方法开发过程中追踪所有化合物。开发的方法现在可由分析人员决定是否验证以供进一步使用或转移至另一个实验室。通过制定专用的方法开发策略,例如本文所述的系统性筛选策略,即使是新手或中等水平的分析人员也能够快速有效地开发新方法。
参考资料
- Cole D, Pagano N, Kelly M, Ellingboe J. Use of Design of Experiments to Optimize High-Throughput Semipreparative LC and LC-MS Methods. J. Comb.Chem. 2004; 6, 78–82.
- Bohm D, Stachel C, Gowik P. Validation of a Multi-Residue Method for the Determination of Several Antibiotic Groups in Honey by LC-MS/MS.Analytical and Bioanalytical Chemistry.2012; 403, 2943–2953.
- Alkhateeb F, Rainville P. 基于“分析方法质量源于设计”的理念开发缬沙坦和亚硝胺杂质的UPLC-MS分析方法.沃特世应用纪要, 720007033ZH, 2020.
- Alkhateeb F, Rainville P. 基于“分析方法质量源于设计”理念开发使用Arc Premier MaxPeak高性能表面(HPS)技术分析磷酸地塞米松及相关化合物的方法.沃特世应用纪要, 720007272ZH, 2021.
- Johansson E, Karlsson A, Ludvigsson J. Ultra High Performance Liquid Chromatography Method Development for Separation of Omeprazole and Related Substances on Core-Shell Columns Using a Quality by Design Approach.Journal of Separation Science. 2020 Feb; 696–707.
- Maziarz M, McCarthy S, Wrona M. 使用系统性筛选策略提高方法开发效率.沃特世应用纪要, 720005026ZH, 2014.
- Durkin MJ, Jafarzadeh SR, Hsueh K, Sallah YH, Munshi KD, Henderson RR, Fraser VJ.Outpatient Antibiotic Prescription Trends in the United States: A National Cohort Study. Infect Control Hosp Epidemiol.2018 May;39(5):584–589.
720007352ZH,2021年8月