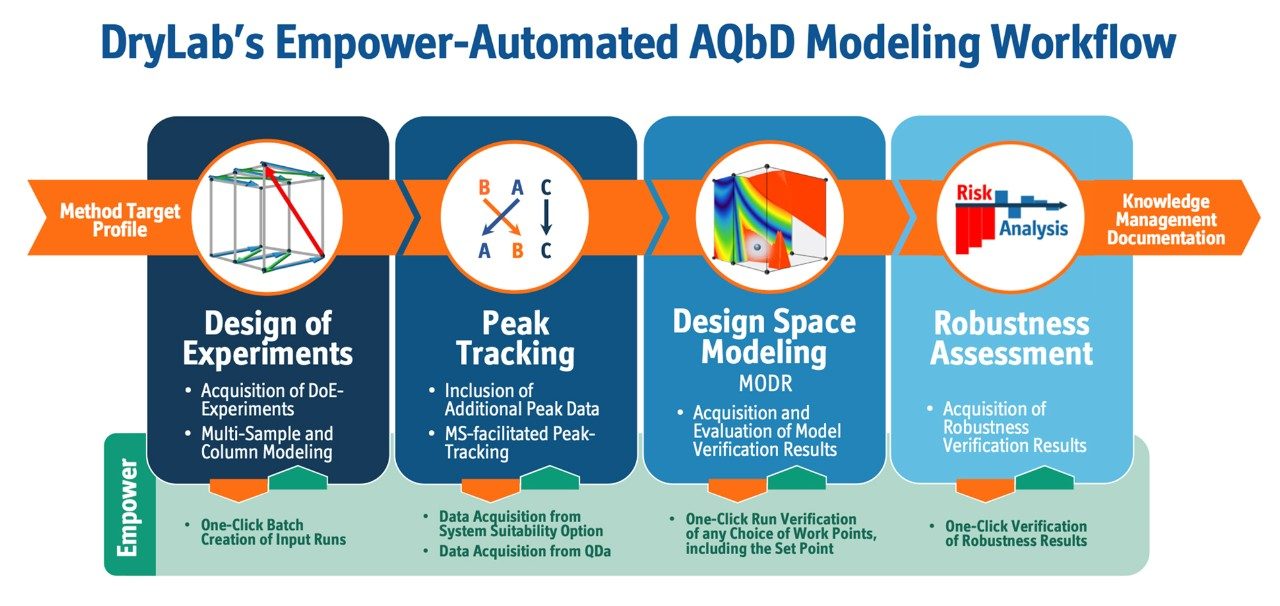
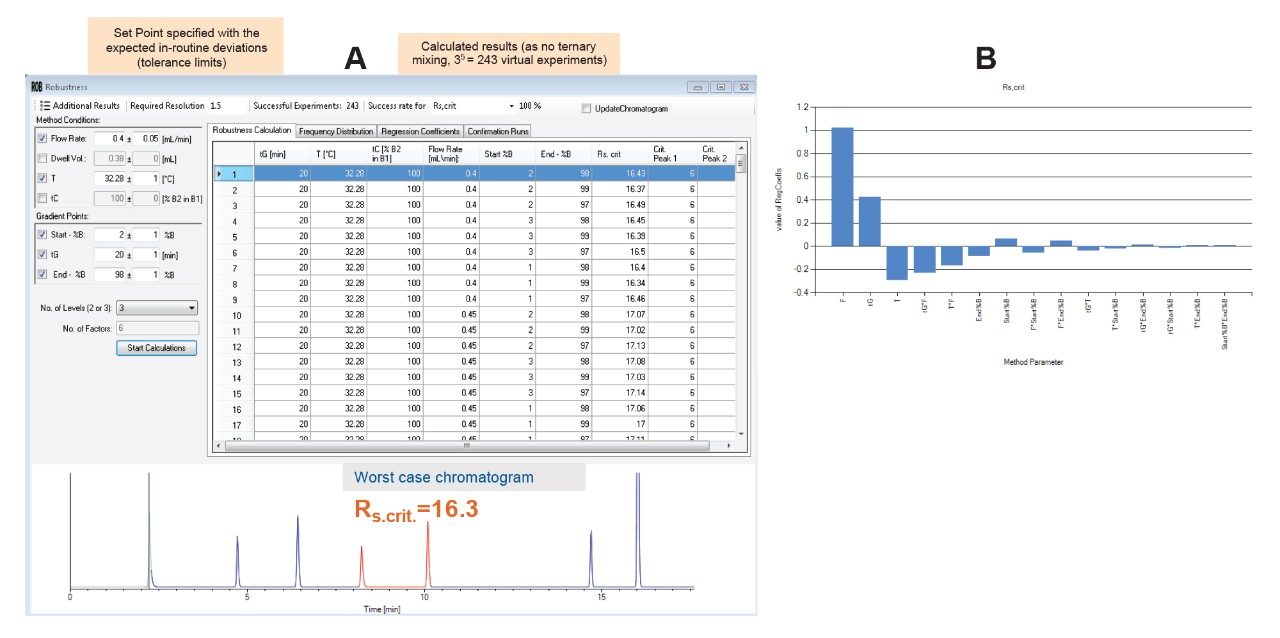
Genotoxic impurities are defined by the ICH S2 (R1) guideline as substances that have been demonstrated to cause deleterious changes to the genetic material regardless of the mechanism.1 The primary concern related to such impurities that they have the potential to interact with human cells to cause mutations and cancer, even at the lowest levels. As such, genotoxic impurities should be avoided all at once and if not possible, reduced below a defined level. The advent of these impurities in various angiotensin receptor blocker drugs (ARB) such as valsartan, losartan, and irbesartan has caused broad recalls of these drugs from the markets and made this issue a focus for regulatory agencies including the FDA and the European Medicines Agency (EMA). In order to comply with regulations, it is critical that analytical methods that are used for the analysis of these compounds are accurate, robust, and sensitive. One way to develop robust and accurate methods is employing the Analytical Quality by Design principles (AQbD) in the method development process. The AQbD is a systematic approach for method development that starts with predefined objectives and provides rational understanding of the effects of chromatographic factors on the method performance.2 In this approach multiple variables are screened to provide a broad knowledge about the impact of the studied factors on the method performance. This knowledge is used to establish the method operable design region (MODR) which corresponds to the multi-dimensional combination of factors that have been verified to meet the method performance criteria. The outcome of this approach is a fit for purpose, well designed, understood, and robust method that delivers the expected performance throughout its lifecycle.3,4
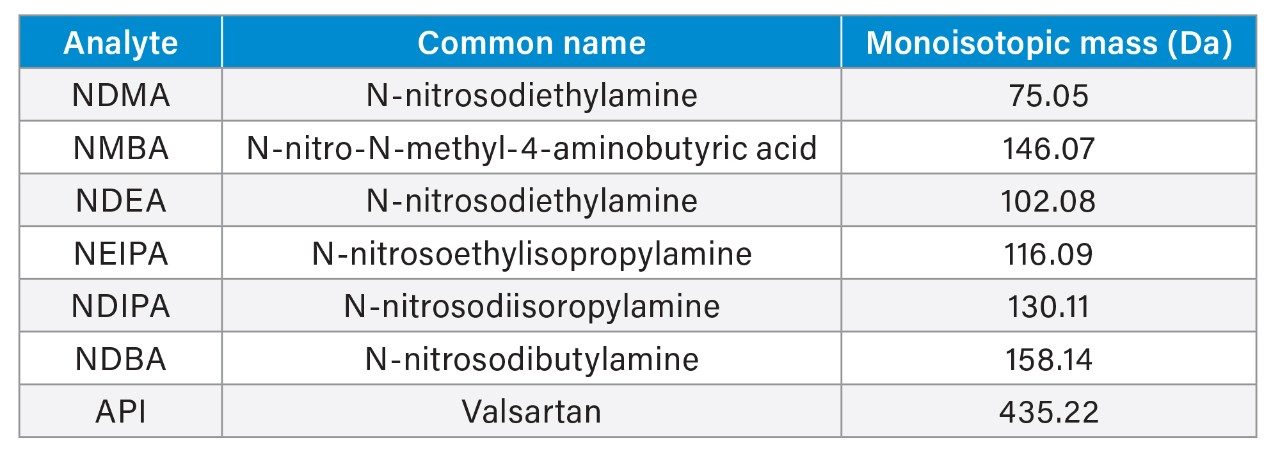
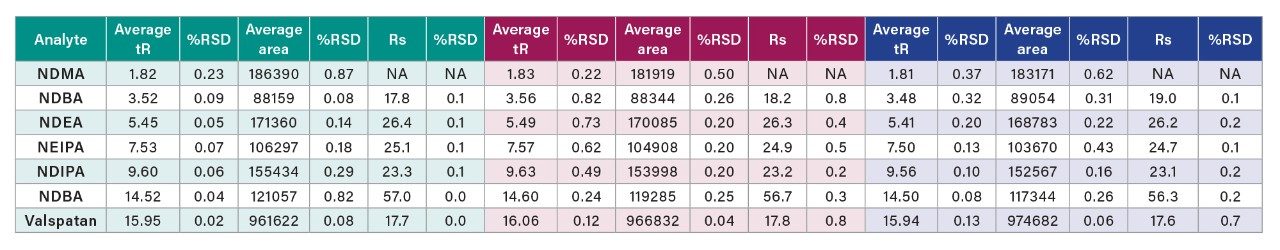
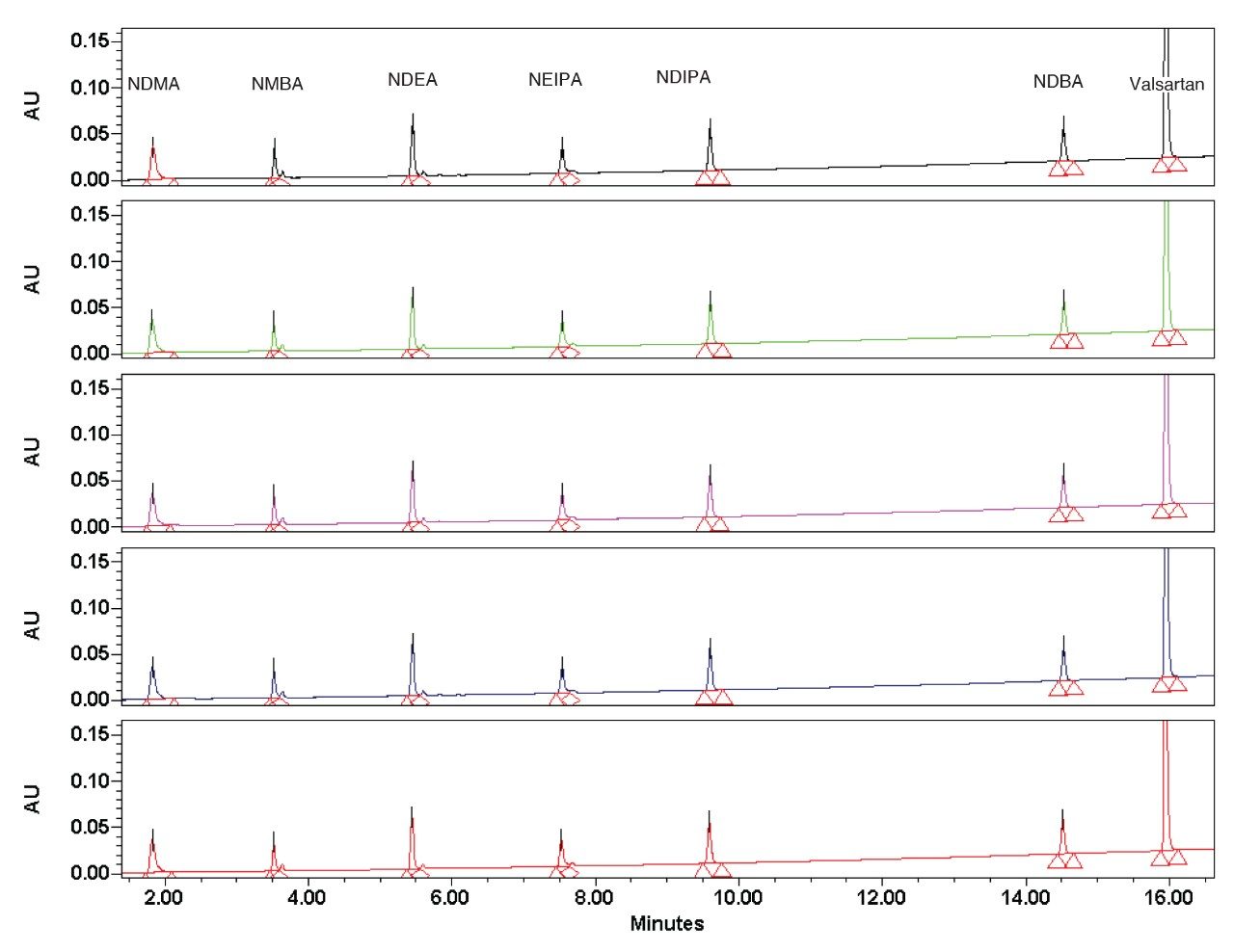
In this application note, a software assisted AQbD approach was implemented to develop a method for the analysis of valsartan standard and six genotoxic impurities (NDMA, NMBA, NDEA, NEIPA, NDIPA, and NDBA).
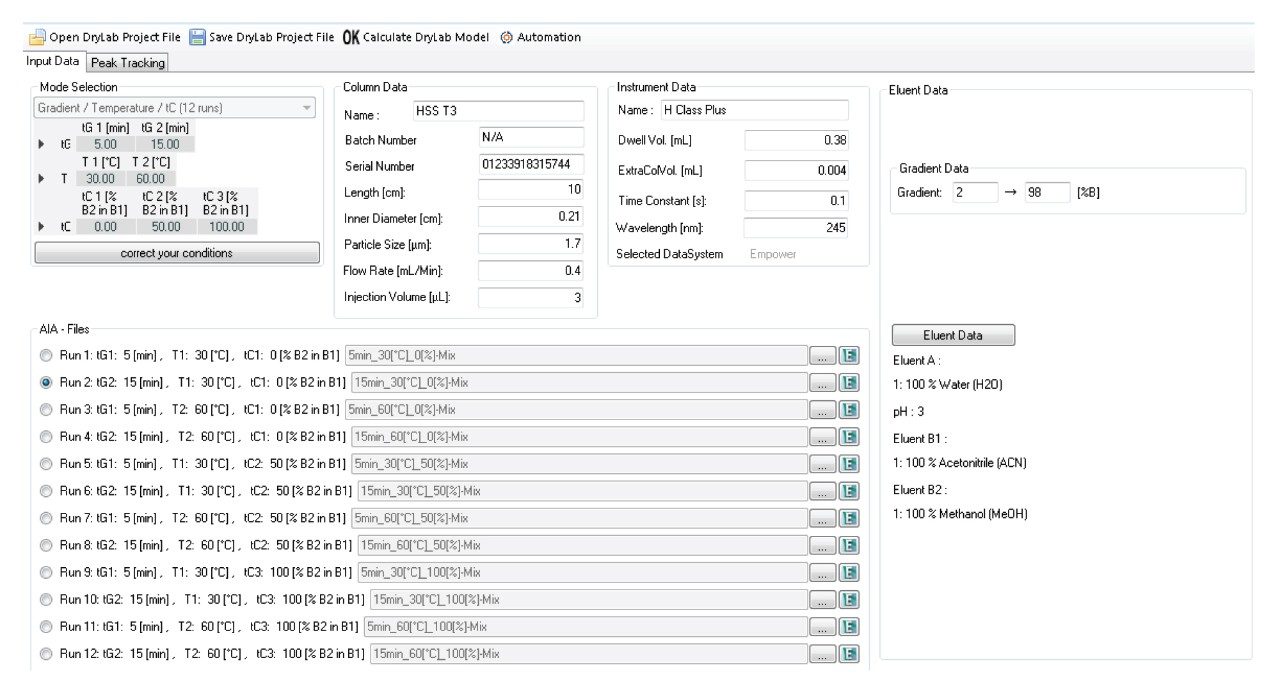
The experiments were performed using an ACQUITY UPLC H-Class System that is equipped with a column manager and solvent select valve to allow for automated exploration of a wide range of conditions.
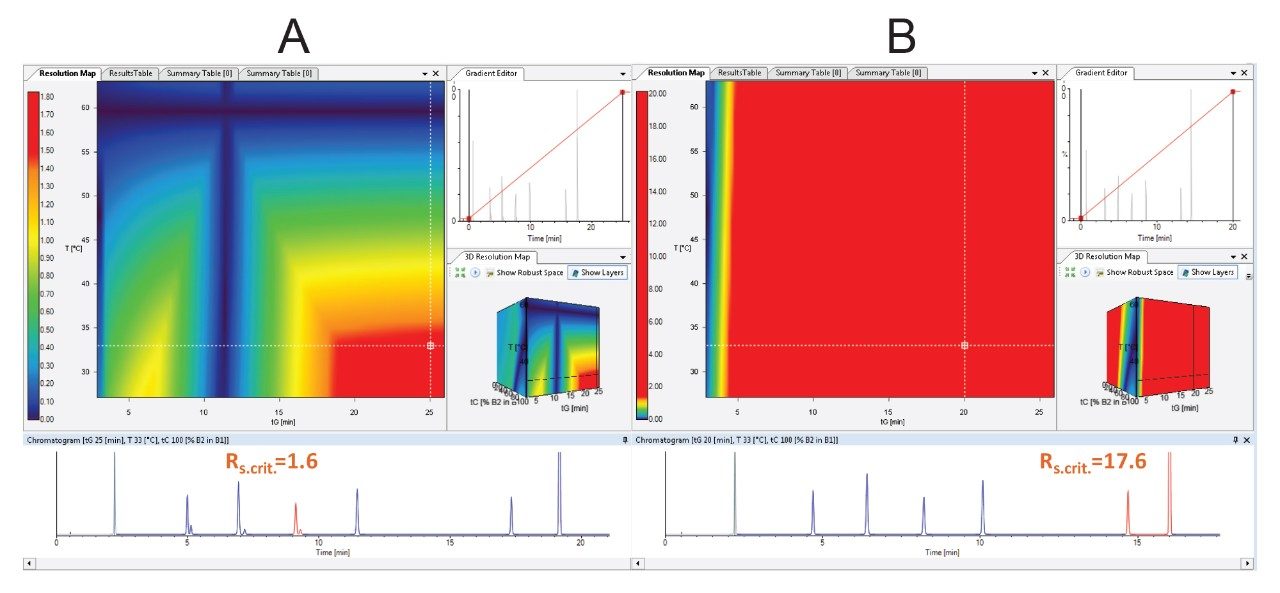
DryLab4 method development software was used as an AQbD software in this study. Details about the use of the AQbD principles for method development on an ACQUITY UPLC H-Class PLUS System has previously been described.5