Tandem Quadrupole Acquisition Modes in DMPK Studies
Abstract
Tandem quadrupole mass spectrometers are extremely powerful and flexible instruments. They are the instruments of choice for high sensitivity quantitative analysis, with detection limits at the pg/mL level readily attainable and thus are extensively used to support DMPK studies such as first time in human (FTIH). The configuration of tandem quadrupole instruments along with modern fast data acquisition facilitates multiple other modes of data acquisition, such as neutral loss, precursor ion scanning, product ion scanning and polarity switching, allowing the DMPK scientist to easily and quickly interrogate their samples to aid the drug discovery or regulator submission process.
Benefits
Introduction
Tandem quadrupole mass spectrometers are the workhorse of quantitative LC-MS/MS analysis due to their selectivity, specificity, and sensitivity. This makes then ideal for the analysis of trace levels of target analyte in complex mixtures such as biofluids, wastewater, formulation, and food stuffs, etc.1 In the pharmaceutical industry tandem quadrupole instruments are used in manufacturing to measure trace levels of impurities in drug substance and drug product. They are also extensively used for the quantification of drug and their metabolites in biological fluids to support discovery, preclinical development, and clinical development studies.2,3 The exquisite sensitivity of multiple reaction monitoring mode (MRM) allows target analytes to be routinely measured in the pg/mL or even fg/mL range with a wide dynamic range of 4–5 orders possible.4 The fast data acquisition speeds of modern tandem quadrupole instruments make them highly suited to coupling with high resolution chromatography methods such as capillary GC or UHPLC where peaks widths of 1–2 seconds are possible. As a result of these attributes tandem quadrupole MS coupled with liquid chromatography operating in multiple reaction monitoring mode (MRM) has become the technology of choice to support drug discovery and development DMPK studies.
Along with the quantification of drugs and their metabolites in biofluids, such as urine, plasma, serum, tissues, bile, cerebrospinal fluid (CSF), etc., there is also a need to interrogate these samples for the presence of drug related metabolites; to ensure toxicological coverage, detect new metabolites and monitor for biotransformation which could be indicators of drug related toxicity such as glutathione’s.5,6 The unique configuration and ion optics of tandem quadrupole MS facilitates multiple different acquisition modes, such as neutral loss, precursor ion scanning, product ion scanning, and polarity switching as well as other data dependent acquisitions combining two or more of these acquisition modes to be performed. Although these nominal mass acquisition modes cannot be used for definitive structural reporting according to ACS guidelines, they are valuable additional modes of data collection that adds versatility to tandem quadrupole MS instrumentation, such as method development, troubleshooting, and screening, in addition to high sensitivity quantification.
These acquisition modes can be used to monitor for drug related material via diagnostic fragment ions, screen for classes of drug metabolites such as sulphates and glucuronides and for drug related impurities via diagnostic fragment ion. These acquisition modes can also be used to confirm the presence of toxic biotransformations (such as glutathione) via data dependent acquisition (DDA) acquisition modes using product ion scanning to trigger MS/MS acquisitions with or without polarity switching. Here we describe the application of these different acquisition modes in drug metabolism studies and highlight the simplicity and versatility across Waters™ Tandem Quadrupole instruments.
Experimental
Sample Description
Methapyrilene and its metabolites in rat urine were obtained from a 5-day repeat dose IV in the male wistar rat. Methapyrilene was administered intravenously at 150 mg/Kg. During sample collection the animals were housed singularly in dedicated metabowls. The urine samples were collected on ice and stored frozen at -20°C prior to analysis. The samples were prepared by mixing 50 µL of urine with 200 µL ice cold acetonitrile, the sample was vortex mixed then stored at -20°C for 1-h. The resulting samples were then centrifuged at 9,000 g for 5 min and the supernatant transferred to a total recovery vial for analysis by LC/MS/MS. Blood was collected via vena cava at the terminal collection into Minivette POCT HeLi coated capillaries 24-h post dose on D2, 4, and 6. Rat plasma samples (50 µL) were prepared by protein precipitation with 200 µL of acetonitrile : methanol (90:10) after vortex mixing the samples were centrifuged at 25000 g for 5 mins. 1 µL of extract was injected onto the chromatography system for analysis by UPLC-MS/MS.
The rats included in the study were housed at Evotec France SAS Animal facility. This facility is accredited by the French Ministry of Agriculture and by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC). This study is compliant with the corresponding project APAFIS #32640–2021101419119467 v5. This project was reviewed by Evotec France Ethical Committee (identified below as CEPAL: CE 029) and authorized by the French Ministry of Education, Advanced Studies and Research.
Method Conditions
The urine samples were screened by the injection of a 2 µL aliquot of sample onto a 2.1 x 100 mm Cortecs™ C8 2.7 µm Column. The column was maintained at 40 °C and eluted with a linear reversed–phase gradient over 10 minutes at 600 µL/min using aqueous 0.1% formic acid as mobile phase solvent A and 0.1% formic acid in 95:5 (v/v) acetonitrile : water as mobile phase B, Table 1. The column effluent was monitored by positive ion ESi mass spectrometry operating in either: i) full scan, ii) neutral loss, iii) precursor ion scanning or iv) product ion scanning mode. The plasma samples were analysed by the injection of a 2 µL aliquot of sample onto a 2.1 x 50 mm Cortecs C8 2.7 µm Column. The column was maintained at 40 °C and eluted with a linear reversed–phase gradient over 2.5 minutes at 600 µL/min using aqueous 0.1% formic acid as mobile phase solvent A and 0.1% formic acid in 95:5 (v/v) acetonitrile : water as mobile phase B Table 2. The column effluent was monitored by positive ion ESi mass spectrometry operating in MRM mode using the transition m/z = 262–119 with a cone voltage of 30 V and a collision energy of 25 eV.
Gradient Table 1 (Urine Analysis)
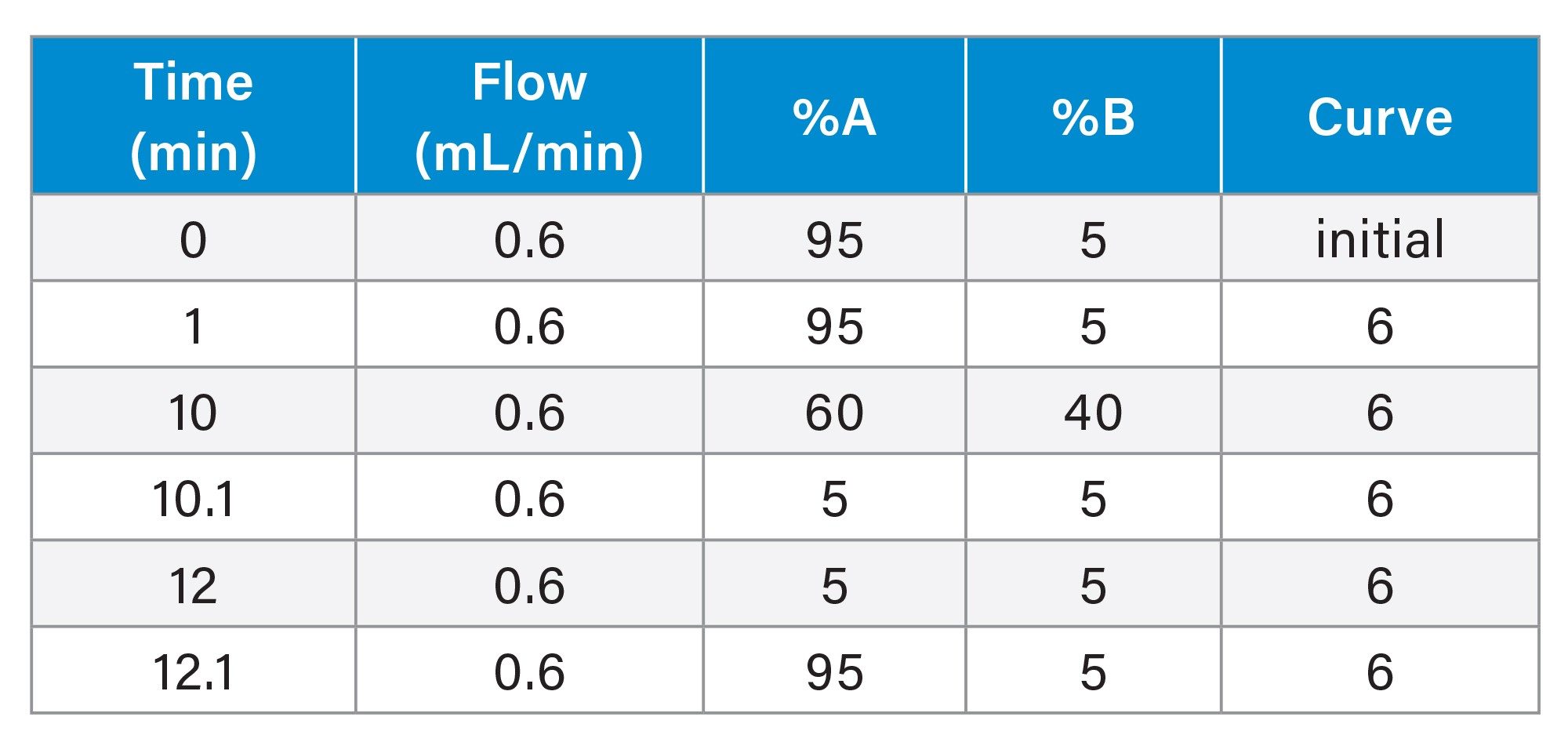
Gradient Table 2 (Plasma Analysis)
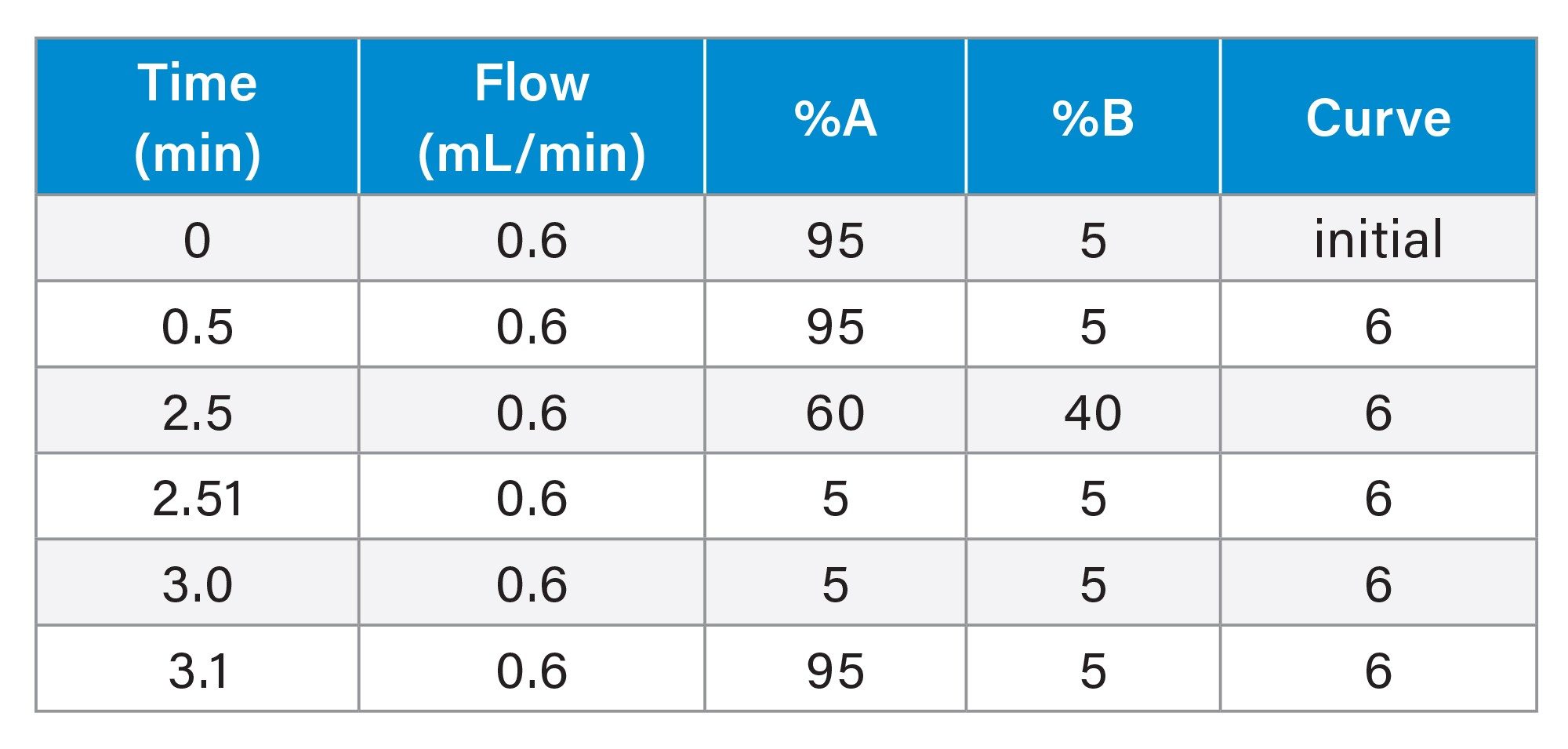
Gradient Table 2 (Urine Analysis)
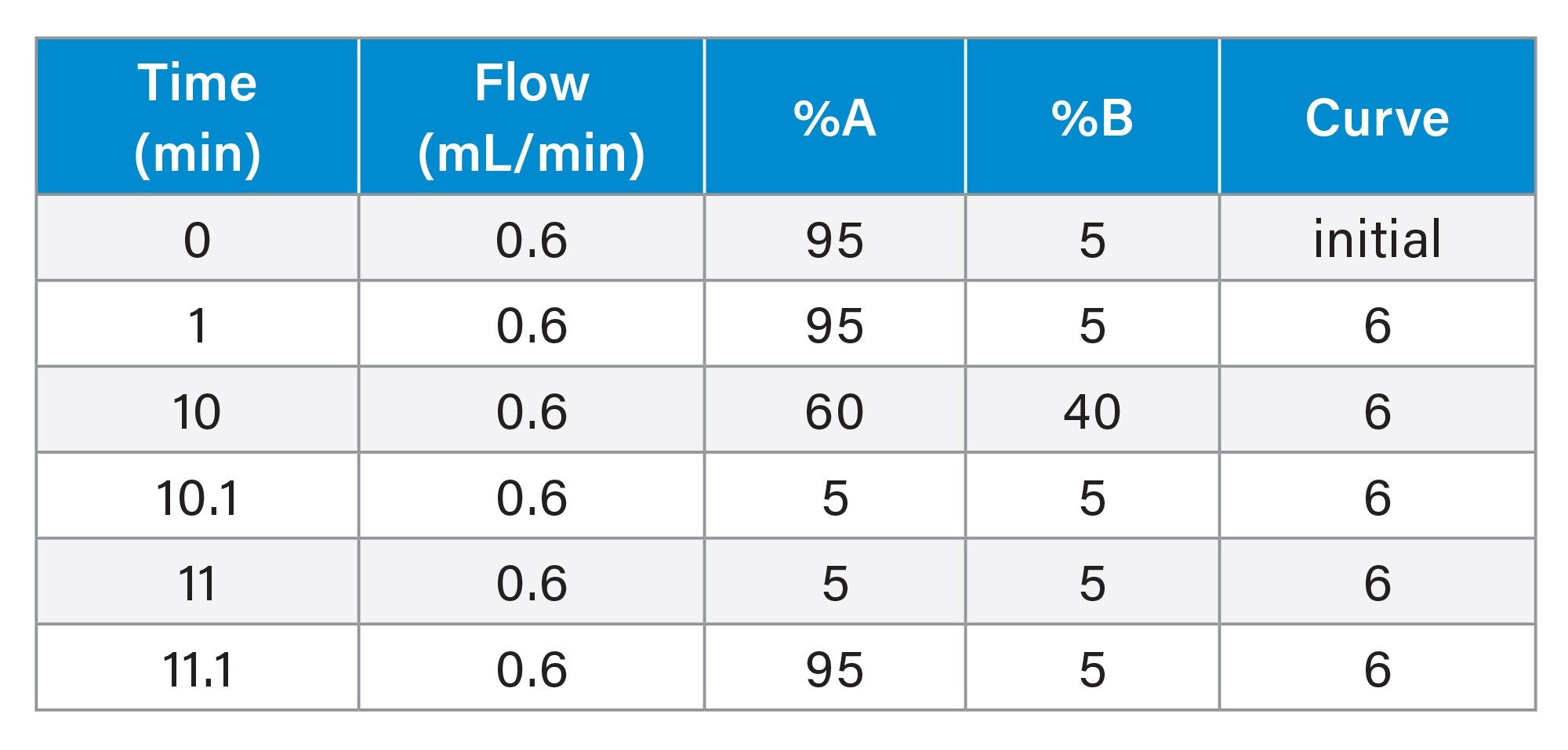
LC Condition
|
LC system: |
ACQUITY™ I Class UPLC™ |
|
Detection: |
Xevo™ TQ-XS |
|
Vials: |
Waters total recovery vials (p/n:186004631) |
|
Column(s): |
CORTECS Premier T3 Column, 2.7 µm, 2.1 x 50 mm (p/n:186010472) or CORTECS Premier T3 Column, 2.7 µm, 2.1 x 100 mm (p/n:186010473) |
|
Column temp.: |
40 °C |
|
Sample temp.: |
8 °C |
|
Injection volume: |
2 µL (urine), 1 µL (plasma) |
|
Flow rate: |
600 µL/min |
|
Mobile phase A: |
0.1% (v/v) aqueous formic acid |
|
Mobile phase B: |
95% acetonitrile, 5% waters, 0.1% (v/v) formic acid |
|
Gradient: |
See gradient table |
MS Conditions
|
MS system: |
Xevo TQ-XS |
|
Ionization mode: |
Positive Ion |
|
Acquisition range: |
ESi |
|
Capillary voltage: |
2.0 Kv |
|
Collision energy: |
30 eV |
|
Cone voltage: |
30 V |
|
Acquisition: |
MRM, Full scan, product ion scan, constant neutral loss, precursor ion scanning |
Data Management
|
Chromatography software: |
MassLynx™ Ver 4.2 |
|
MS software: |
MassLynx Ver 4.2 |
|
Informatics: |
MassLynx Ver 4.2 |
*Note 5. Specify version for each software
Results and Discussion
The Waters Xevo TQ-XS Triple Quadrupole Mass Spectrometers ion path geometry allows for multiple MS acquisition modes to be performed, these include full scan MS(MS), SIR (selected ion recording) /MRM (multiple reaction monitoring), neutral loss, product ion scanning, precursor ion scanning as well as combinations of these acquisition modes, see Table 3. Methapyrilene (Figure 1), an antihistamine and anticholinergic drug, and its metabolites, in rat urine, were used to illustrate the LC-MS results obtained using these various acquisition modes.7
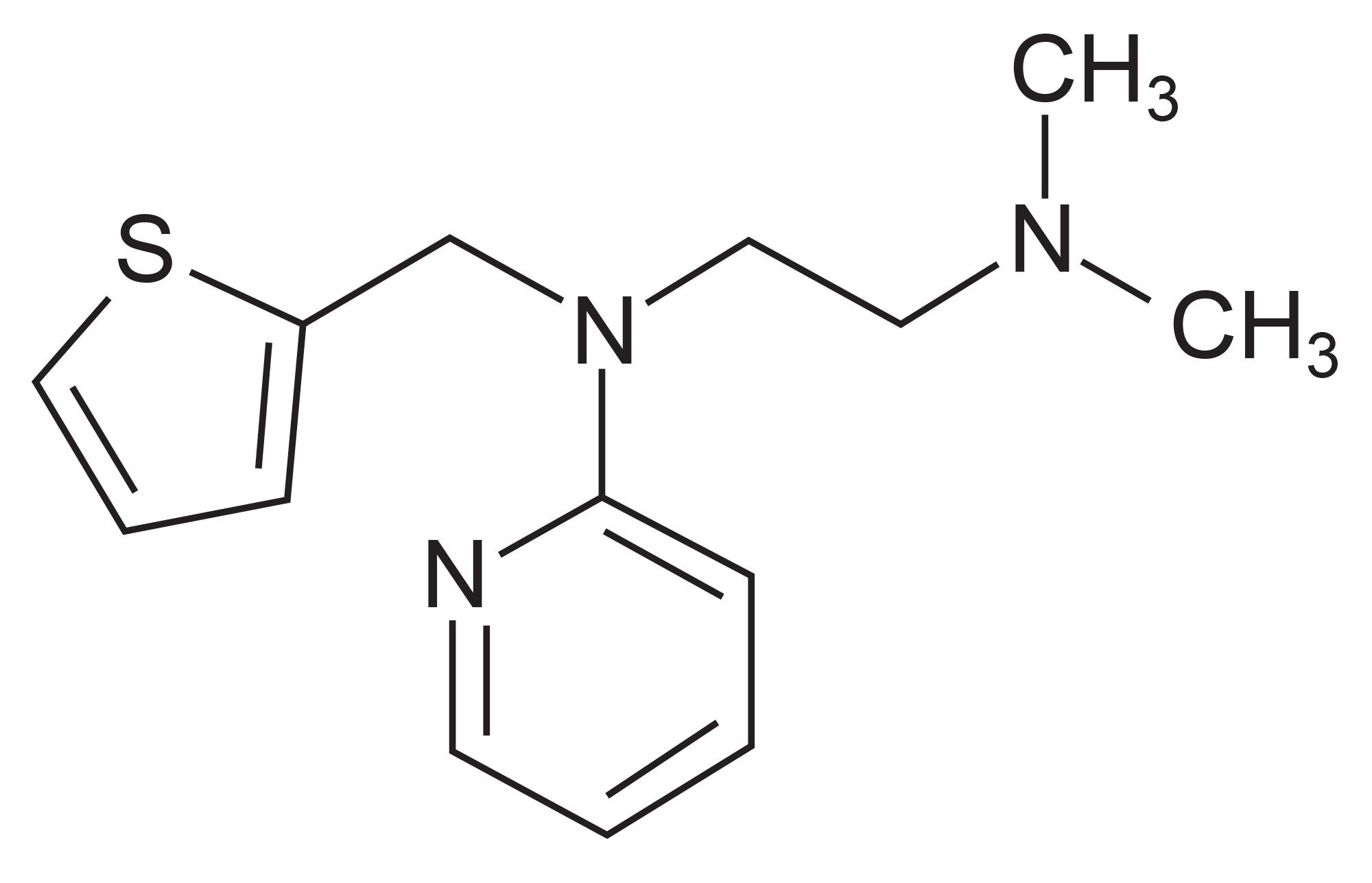 Figure 1. Methapyrilene.
Figure 1. Methapyrilene.
Table 3
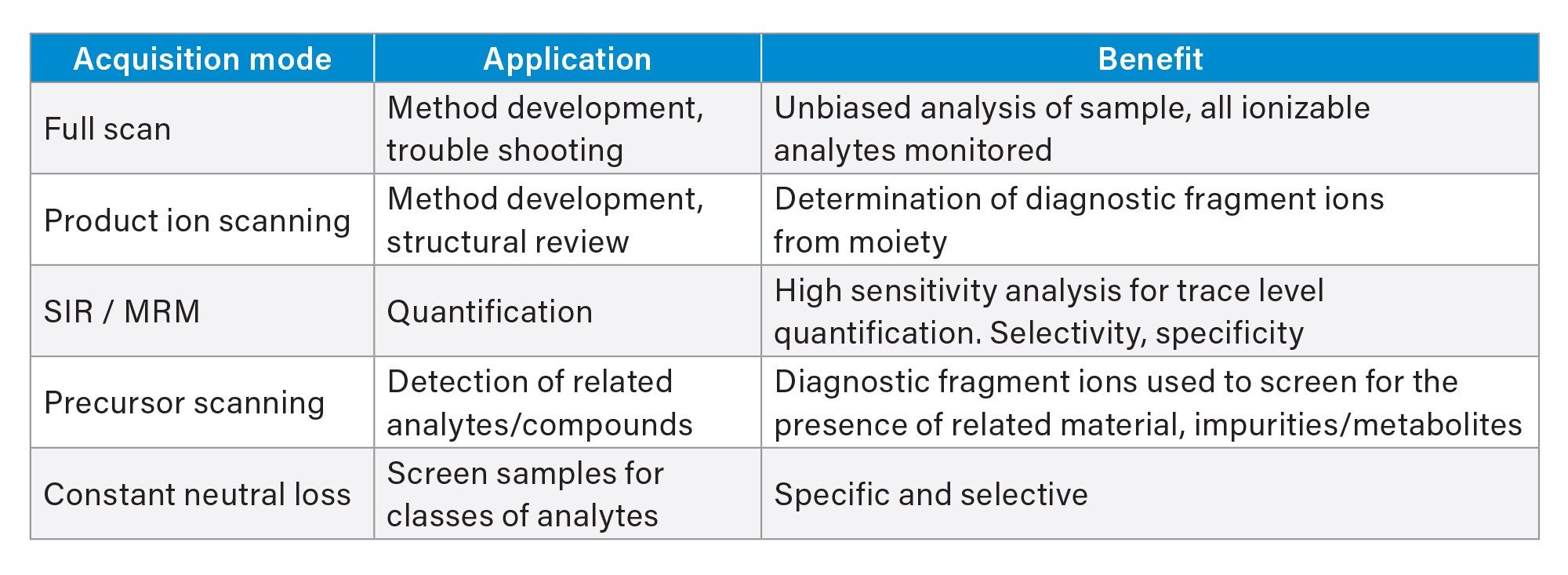
Full scan MS is perhaps the simplest form of MS data acquisition. In this acquisition mode all samples that can be ionized pass through the mass spectrometer, over a pre-set mass range, and signal at the MS detector, Figure 2. The m/z value is determined by the scanning of one of the quadrupoles (over a pre set range e.g., m/z 50–800), such that at any moment in time only ions of a specific m/z reach the detector. The magnitude of the response of each analyte will depend upon its concentration, ability to accept a charge, ionization efficiency, solvent pH, buffer concentration, organic solvent composition etc. In this mode of analysis, it is possible to detect both precursor and product ions for a chemical species. This acquisition mode can be performed in both -ve and +ve ion mode in the same analysis. The data shown in Figure 3 illustrates the LC-MS positive ion chromatogram and MS spectrum obtained from the analysis of the authentic standard of methapyrilene at a concentration of 100 ng/mL, eluting at tR = 2.45 min. Here we can see that the base peak has a mass of m/z = 262.23 and there is also evidence of methapyrilene fragment ions at m/z = 217.14, 166.24 and 96.88. These are likely to have resulted from in source fragmentation during the ionization process.
 Figure 2. Full scan acquisition mode.
Figure 2. Full scan acquisition mode.
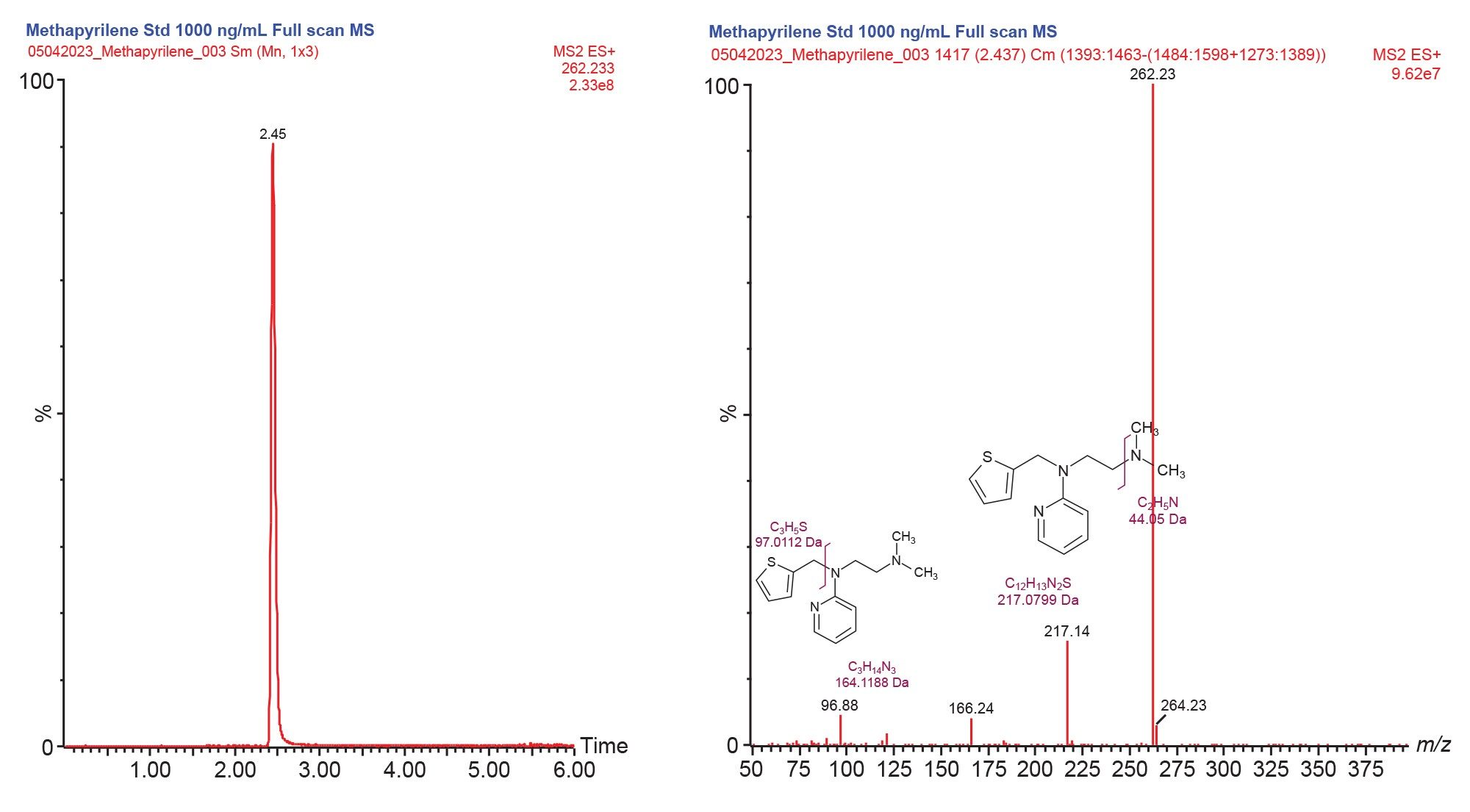 Figure 3. LC-MS chromatogram and full scan MS analysis of methapyrilene authentic standard at a concentration of 100 ng/mL.
Figure 3. LC-MS chromatogram and full scan MS analysis of methapyrilene authentic standard at a concentration of 100 ng/mL.
Full Scan MS/MS
Full scan MS/MS (product ion scanning) data can also be acquired following preselection of a specific ion or m/z value in the first resolving quadrupole, this ion is then directed to the collision cell. The ion is accelerated by the application of an electric potential and then allowed to collide with a neutral gas, such as argon, Figure 4, to produce structural fragments. The resulting fragment ions are then scanned by the final resolving quadrupole and signal is produced by the MS detector. An example of the fragmentation spectra produced for the methapyrilene peak eluting at tR = 2.45 min is given in Figure 5, using m/z = 262.23 as the selected mass in MS1. Here we can see that the m/z = 262.23 ion gave rise to major product ions m/z = 217.09, 133.07, 121.08, and 119. This mode of data acquisition can be used to provide specific fragmentation data to confirm the identity of a particular compound by comparison with an authentic standard.
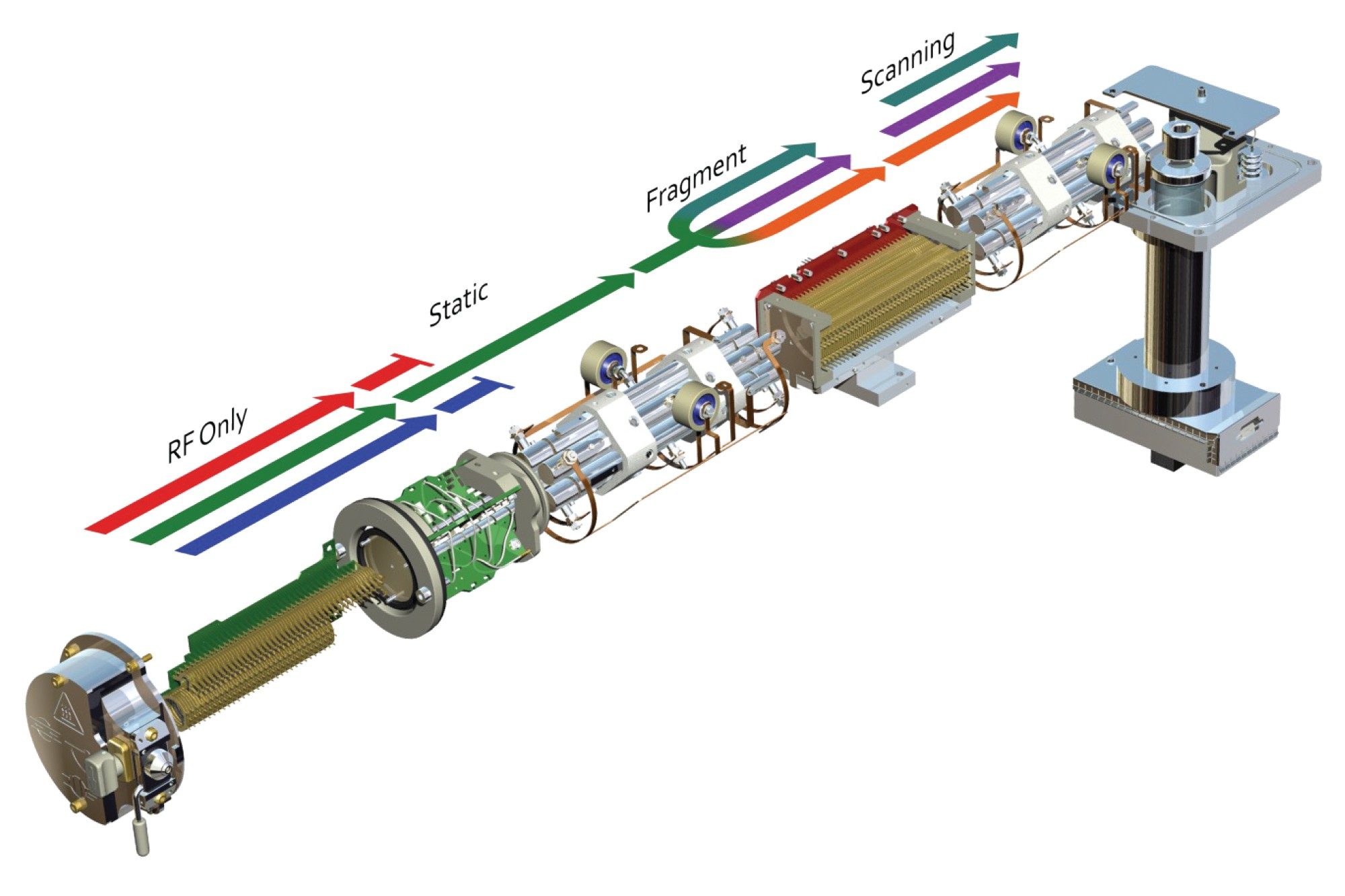 Figure 4. Product ion Scanning mode.
Figure 4. Product ion Scanning mode.
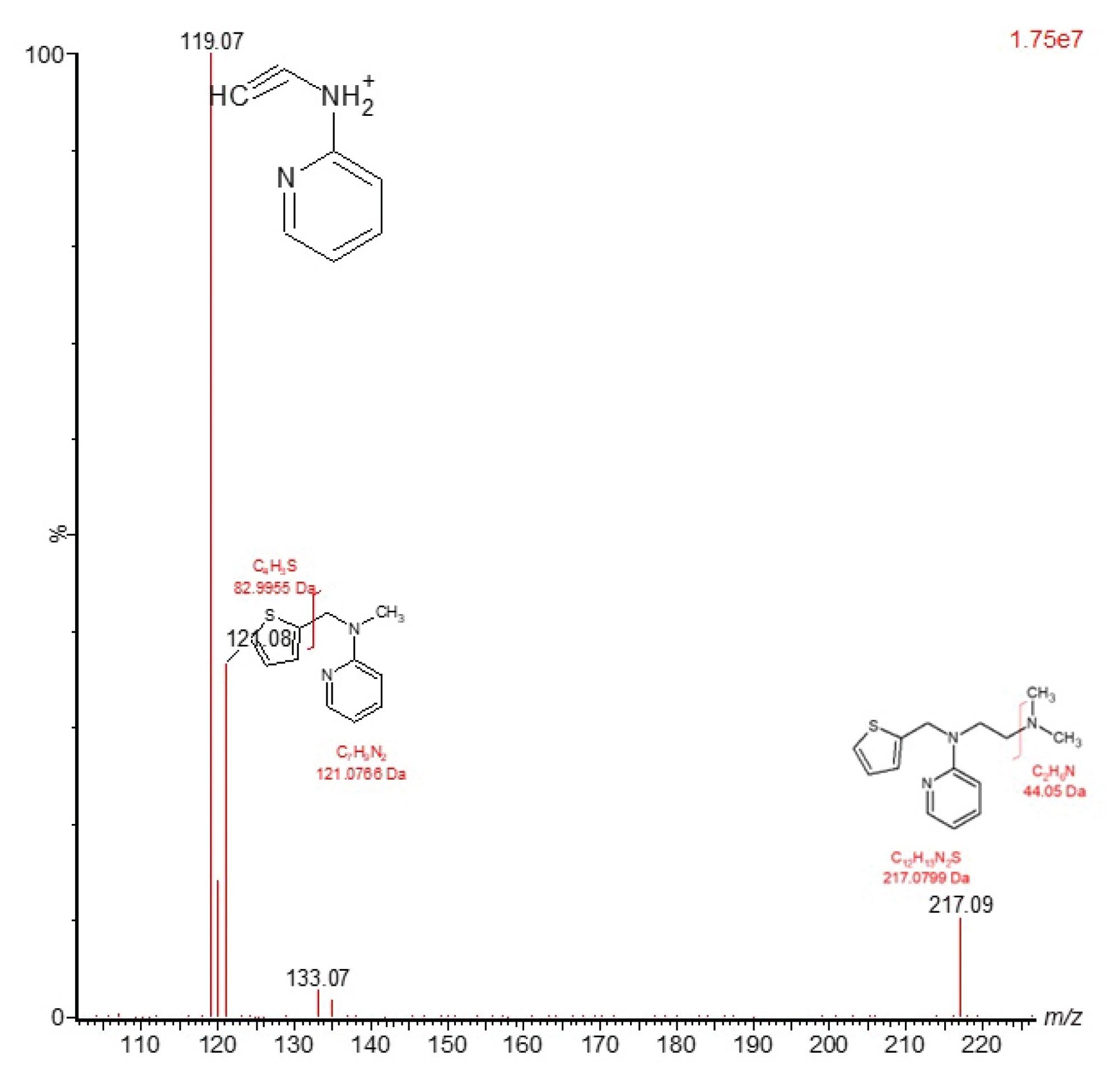 Figure 5. Product ion MS/MS analysis of methapyrilene authentic standard, m/z = 262.23 at a concentration of 100 ng/mL, eluting with a retention time of tR = 2.45 min.
Figure 5. Product ion MS/MS analysis of methapyrilene authentic standard, m/z = 262.23 at a concentration of 100 ng/mL, eluting with a retention time of tR = 2.45 min.
SIR and MRM
The predominant use of Tandem, or Triple, quadrupole mass spectrometers is for quantitative analysis operated in SIR or MRM mode. This is due to their selectivity and specificity, which give rise to exquisite sensitivity. In these modes of analysis only a single or selection of a few ions are allowed to pass through the mass spectrometer and reach the detector for analysis. In selected ion recording (SIR) mode, all the ions are allowed to pass through the first resolving quadrupole and collision cell, then a single ion or small number of intact (i.e., not fragmented) ions are selected in the final resolving quadrupole for measurement at the detector, Figure 6A. In MRM mode a single ion is selected in the first resolving quadrupole, this ion is then directed to the collision cell for fragmentation and a specific product ion selected in the final quadrupole for measurement at the detector, Figure 6B. Positive and negative ion MRM or SIR data collection can be performed in the same acquisition. Due to the specificity of monitoring a precursor to fragment reaction, multiple reaction monitoring (MRM) analysis is the most common mode of analysis used in environmental science and drug metabolism studies, where there is a need to measure extremely low levels of a target analyte(s) in a complex mixture such as foodstuffs or biofluids. An example of MRM analysis for the measurement of methapyrilene in rat plasma, using a rapid RPLC gradient, at a concentration of 1 ng/mL is given in Figure 7. In this data we can see the ability to easily detect the 1 ng/mL sample whilst the blank plasma sample shows no presence of the dug at the same retention time (tR = 1.69 min).
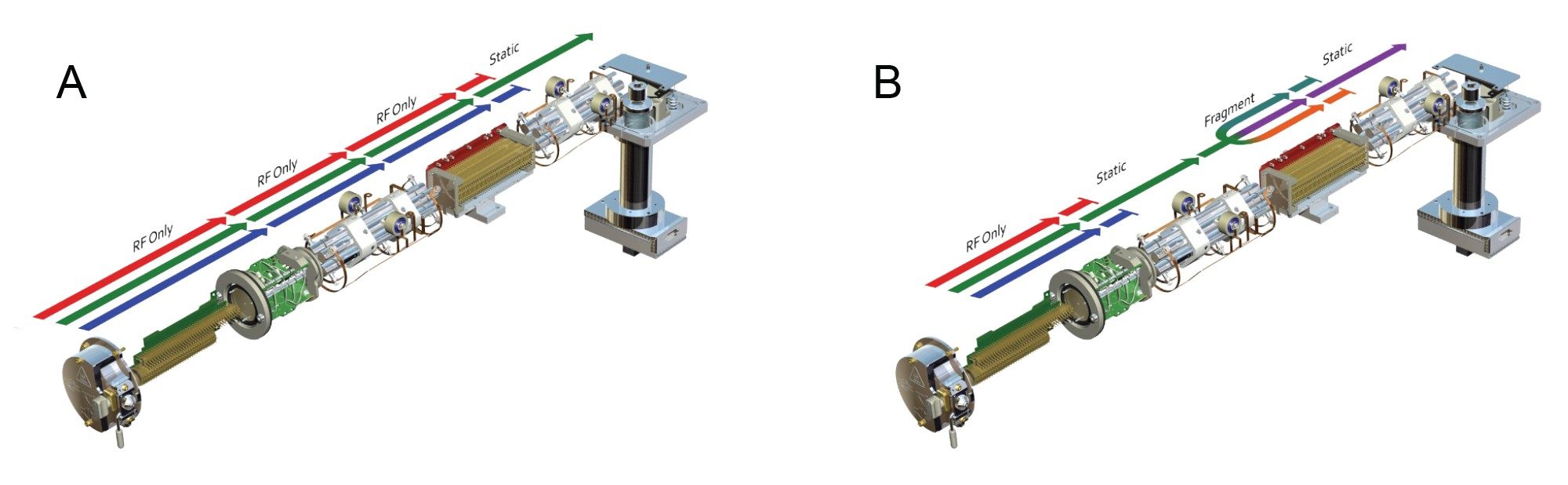 Figure 6. Single Ion Recording (SIR) (A) and Multiple Reaction Monitoring (MRM) (B).
Figure 6. Single Ion Recording (SIR) (A) and Multiple Reaction Monitoring (MRM) (B).
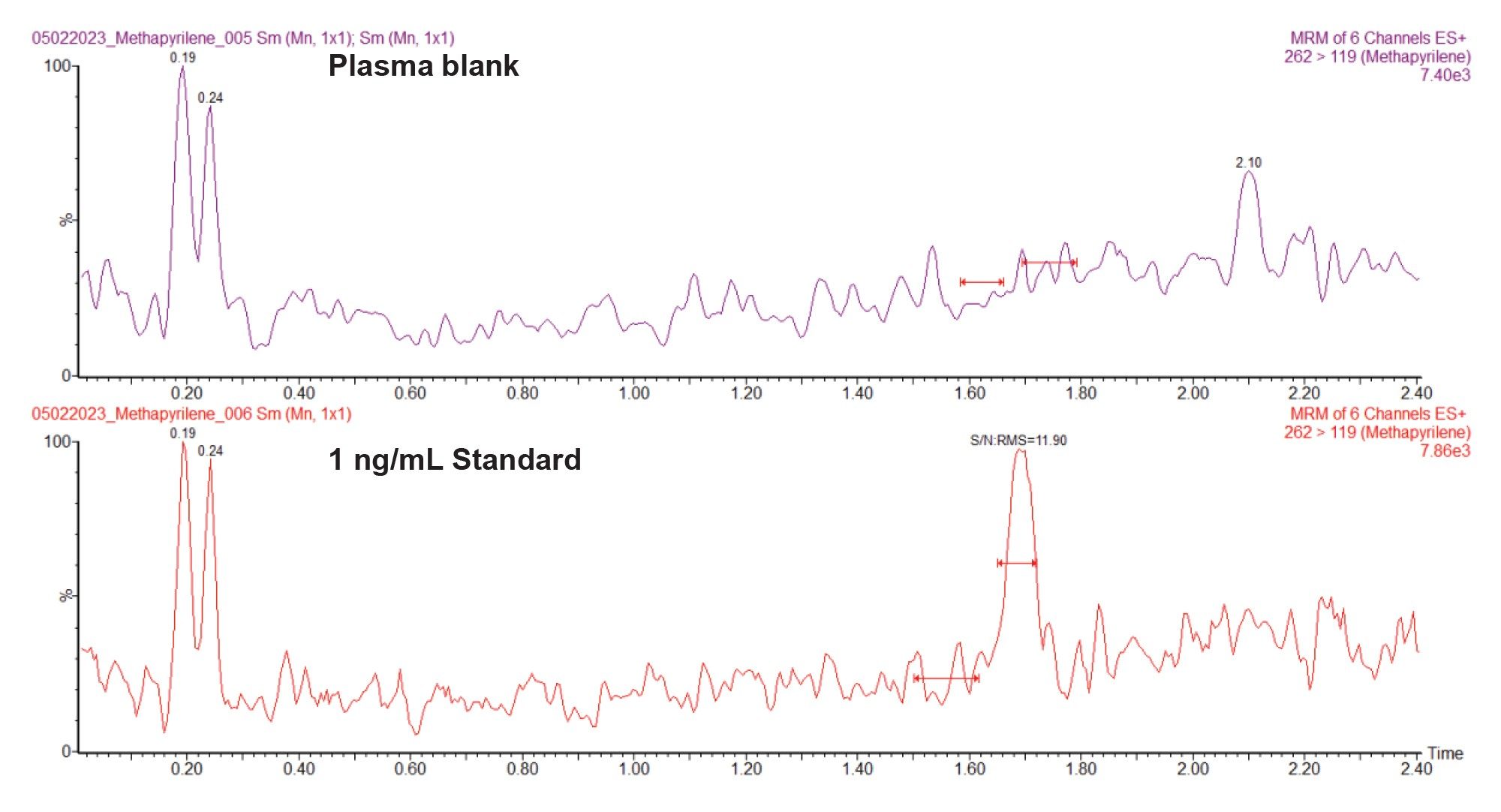 Figure 7. Analysis of methapyrilene in protein precipitated rat plasma, using UHPLC-MS/MS MRM monitoring (262–119) in positive ion mode at a concentration of 1 ng/mL and blank extracted rat plasma.
Figure 7. Analysis of methapyrilene in protein precipitated rat plasma, using UHPLC-MS/MS MRM monitoring (262–119) in positive ion mode at a concentration of 1 ng/mL and blank extracted rat plasma.
Precursor Ion Scanning
Precursor ion scanning enables the identification of all specific precursor masses in a sample that generate a common (and predefined) fragment ion. Precursor ion scanning utilizes a combination of scanning, fragmentation and selected ion recording. In this mode of analysis, the first resolving quadrupole is set to scan a across specific mass range. As the ions exit Q1 they are directed to the collision cell where they are fragmented using either a single collision energy or a collision energy ramp. The fragment ions exiting the collision cell are then filtered by the final resolving quadrupole (Q3) which is operated in a static mode allowing only ions of a specific (and predefined) mass-to-charge ratio to pass through, Figure 8. Only analytes which possess a fragment with ion specific m/z value are detected. The results produced allow the researcher to visualize which precursor ions gave rise to the diagnostic fragment ion selected in Q3.
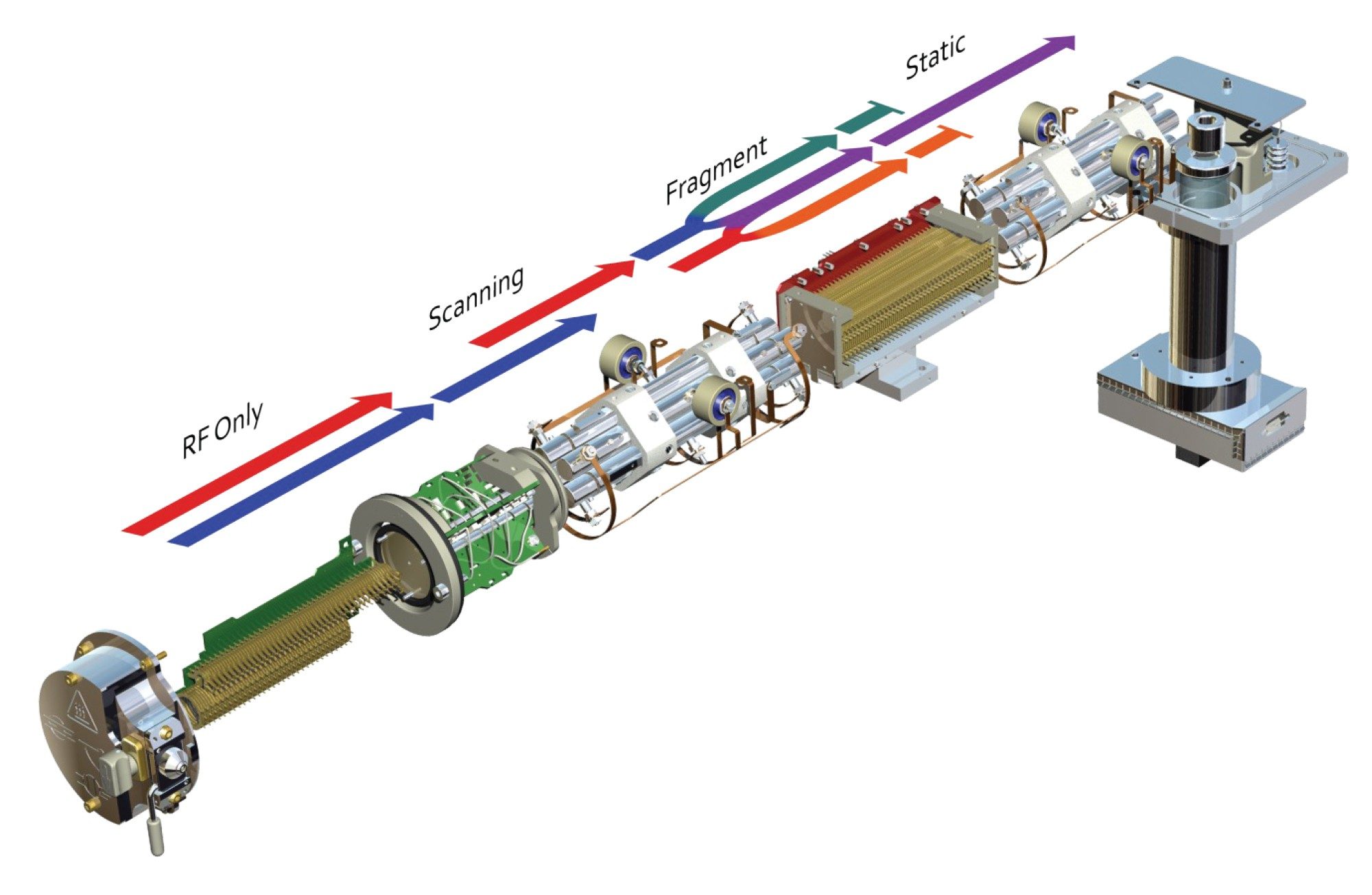 Figure 8. Precursor ion scanning acquisition mode.
Figure 8. Precursor ion scanning acquisition mode.
Precursor ion scanning is used in drug metabolism studies to identify drug related biotransformations in complex matrices such as bile, urine, plasma and faeces. The data displayed in Figure 9 illustrates the analysis of rat urine 24-h post dose following the oral administration of methapyrilene at 150 mg/Kg. The sample was analysed using precursor ion scanning of the fragment ion m/z=96.88 in positive ion mode. The resulting chromatogram (Figure 9 left) illustrates all of precursor ion peaks detected which give rise to the product ion m/z=96.88. In Figure 9 right the MS2 spectrum has been extracted from the peak eluting at retention time tR = 3.04 min. This analyte give rise to a base peak m/z = 294.25, with an insource fragment ion m/z=233.22. This is a common fragment to methylpyrilene and the determined precursor m/z of 294.25 is a +32 Dalton mass shift from the mass of the parent drug indicates that this peak is drug related and is potentially the dihydroxyl metabolite of methapyrilene. Based on the MS spectra shown in Figure 3 and previous publications investigating the metabolic fate of methapyrilene this metabolite is likely to be the dihydroxyl metabolite of methapyrilene, The m/z=233.22 ion suggests that one of the positions of hydroxylation is on the pyridine-thiol ring fragment, with the other position of hydroxylation being on the tertiary amine group.8
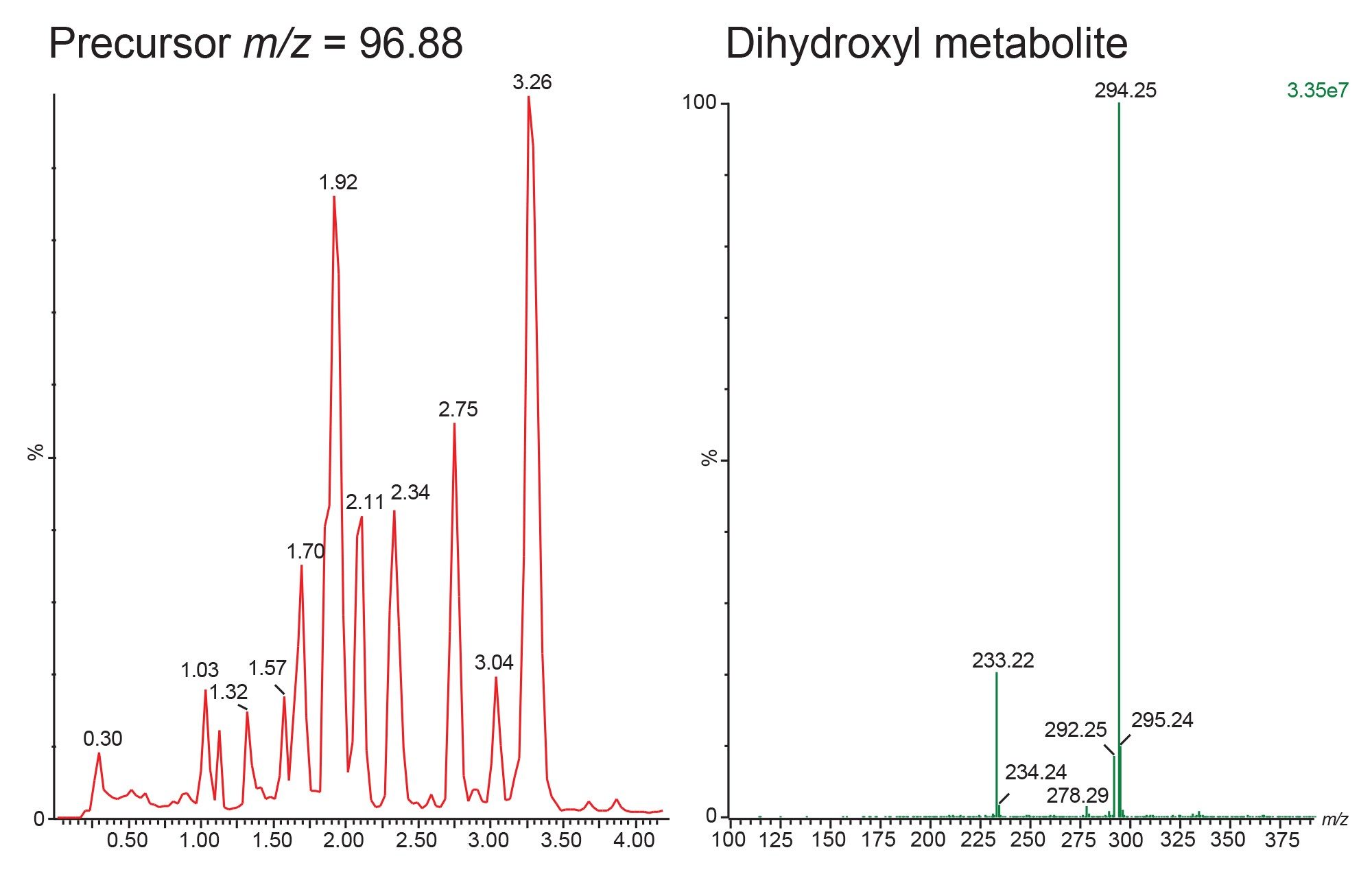 Figure 9. Positive ion LC-MS/MS analysis of rat urine (24-h post dose) following the oral administration of methapyrilene at 150 mg/Kg using precursor ion scanning m/z=97.88 Extracted ion chromatogram on the right illustrates the MS spectrum of the peak eluting at tR=3.04 min.
Figure 9. Positive ion LC-MS/MS analysis of rat urine (24-h post dose) following the oral administration of methapyrilene at 150 mg/Kg using precursor ion scanning m/z=97.88 Extracted ion chromatogram on the right illustrates the MS spectrum of the peak eluting at tR=3.04 min.
Constant Neutral Loss
Constant neutral loss acquisition employs the simultaneous scanning of both the first resolving quadrupole and final resolving quadrupole with fragmentation of the ions in the collision cell at either a pre-set energy or using a collision energy ramp. The two quadrupoles Q1 & Q3 are set to scan whilst keeping a fixed mass difference between them, Figure 10. This acquisition mode can be employed to screen for analytes which have a common structural feature such as sulphate or phosphate groups.
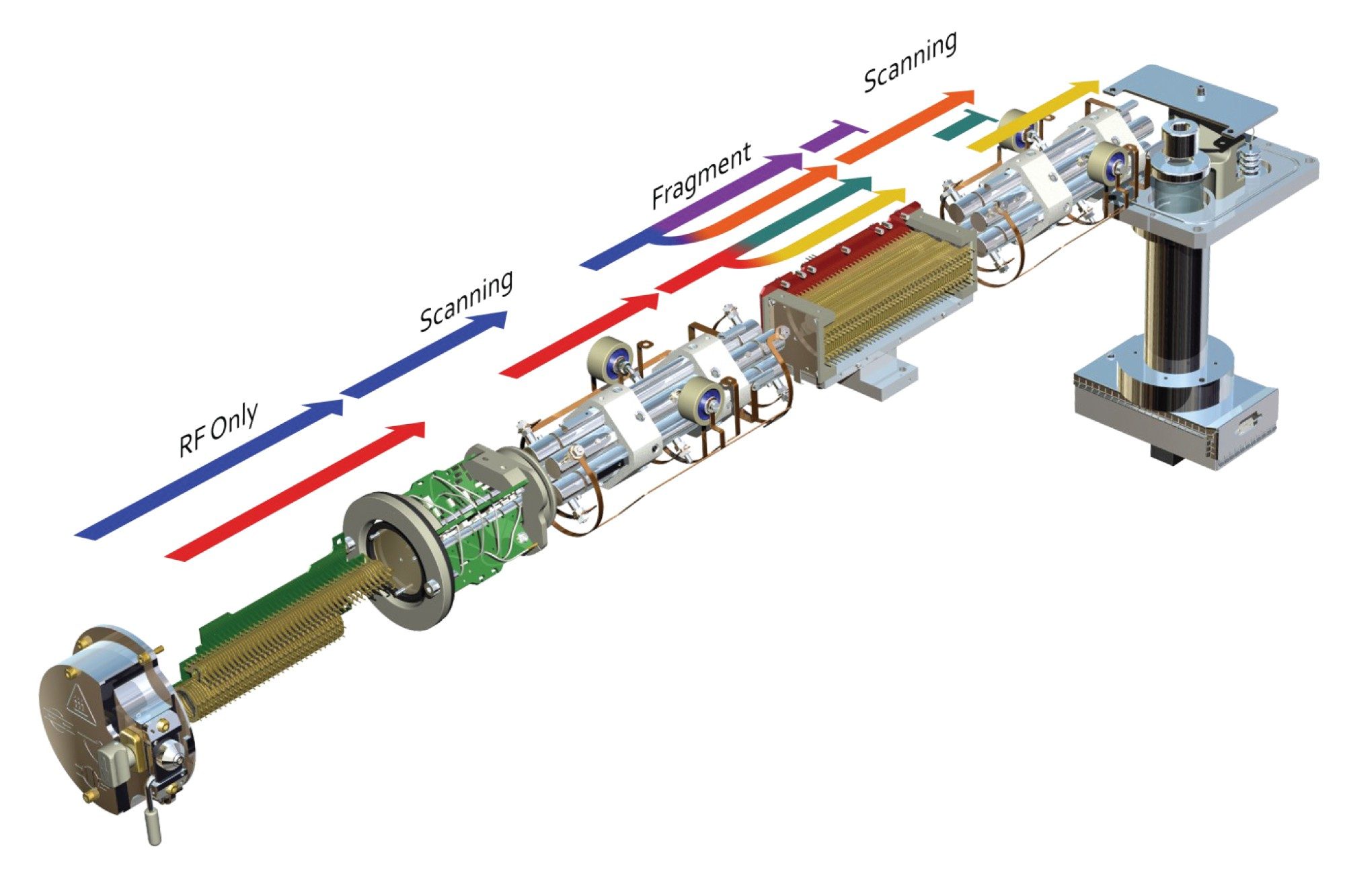 Figure 10. Constant neutral loss acquisition mode.
Figure 10. Constant neutral loss acquisition mode.
Constant neutral loss acquisition mode of analysis has been extensively employed by metabolic researchers to screen for drug conjugates such as sulphates, glucuronides and potentially toxic metabolites such as glutathione’s. Glucuronidation and sulphation are two of the most common routes of conjugation in mammalian drug metabolism, the neutral loss of 176.12 Da, in positive ion mode MS, is characteristic of the loss of the glucuronic acid moiety. The positive ion ESi LC-MS/MS chromatogram obtained from the analysis of rat urine following the oral administration of methapyrilene (150 mg/Kg, D6) using the constant neutral loss of 176.12 Da data is displayed in Figure 11. As can be seen from this data multiple peaks were detected using the neutral loss 176.12 Da, most of which are glucuronide conjugates of endogenous metabolites.
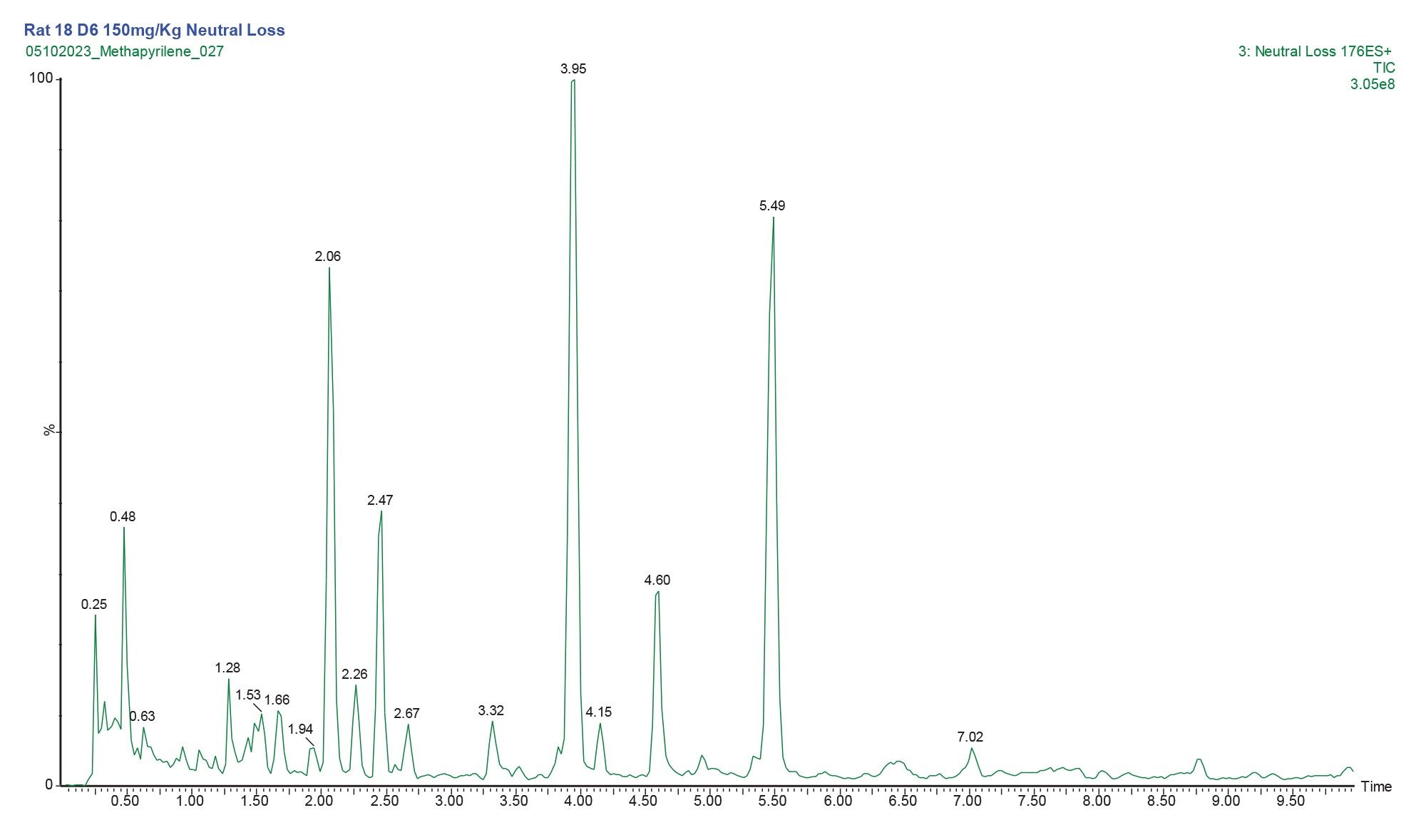 Figure 11. Positive ion LC-MS-MS chromatogram obtained from the analysis of constant neutral loss 176.12 Da of rat urine D6 24-h following the repeat oral administration of methapyrilene at 150 mg/Kg.
Figure 11. Positive ion LC-MS-MS chromatogram obtained from the analysis of constant neutral loss 176.12 Da of rat urine D6 24-h following the repeat oral administration of methapyrilene at 150 mg/Kg.
Previous publications have reported that O-glucuronidation is a major route of elimination of methapyrilene with a signal m/z=454.20. The extracted ion chromatogram displayed in Figure 12A shows the presence of 3 chromatographic peaks which have a precursor mass m/z=454.20 and exhibit the neutral loss of 176.12 Da. The spectrum obtained from the peak eluting at tR=1.91 min shown in Figure 12B, the data shows the presence of a peak at m/z=454.2 as well as peaks at m/z=409.20, 313.20, 310.20, and 245.20.
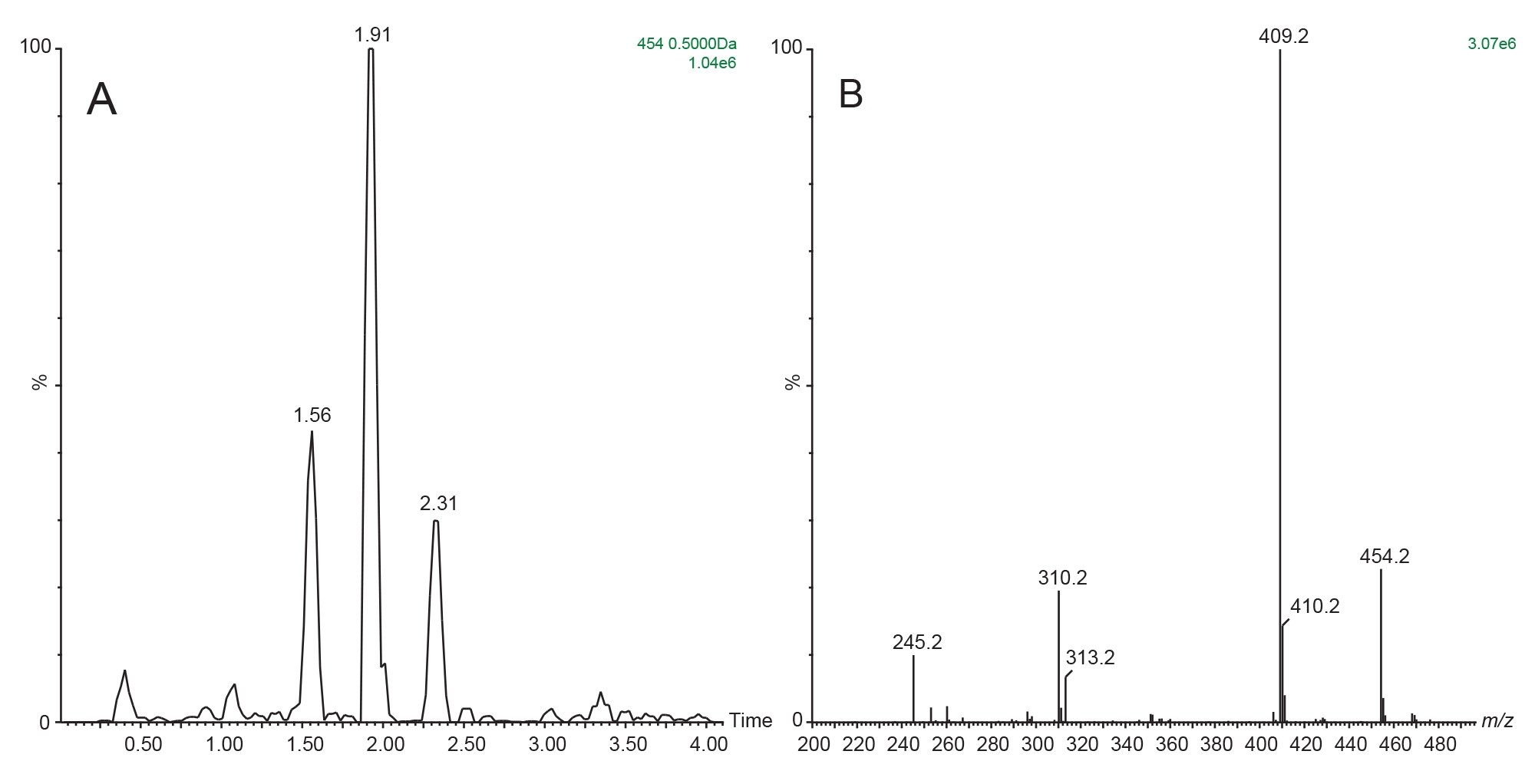 Figure 12. Extracted ion chromatogram m/z=454.20 (+/- 0.5) from the positive ion ESI LC-MS/MS analysis of rat urine D6 24-h post dose following the oral administration of methapyrilene at 150 mg/Kg using the neutral loss 176.12 Da (A). MS spectrum of peak eluting at tR=1.91 min (B).
Figure 12. Extracted ion chromatogram m/z=454.20 (+/- 0.5) from the positive ion ESI LC-MS/MS analysis of rat urine D6 24-h post dose following the oral administration of methapyrilene at 150 mg/Kg using the neutral loss 176.12 Da (A). MS spectrum of peak eluting at tR=1.91 min (B).
Conclusion
DMPK involves the quantification of the candidate medicine in in vitro samples and biological fluids as well as the detection, characterization and often the quantification of its metabolites. In the discovery phase of drug development this requires rapid method development, quantitative bioanalysis, and metabolite screening. In the development phase validated high sensitivity assays are required to support first time into human studies as preclinical studies. The Xevo TQ-XS Tandem (triple) Quadrupole Mass Spectrometer is a highly flexible, sensitive instrument, providing a high sensitivity platform for quantitative analysis using MRM data acquisition. It also offers a wide variety of acquisition modes, such as neutral loss, precursor ion scanning and product ion scanning, allowing researchers to profile their samples for drug related metabolites and potentially toxic metabolites. Here we have demonstrated the use full scan MS and MS/MS to understand the fragmentation pattern of methapyrilene, MRM data acquisition to facilitate the high sensitivity quantification of the drug in plasma and urine, precursor ion scanning, and constant neutral loss acquisition to monitor drug related metabolites in urine. This data illustrates the sensitivity, flexibility, and ease of use of the Waters Xevo TQ-XS for discovery DMPK studies and core laboratory operations.
References
- C Sack, M Smoker, N Chamkasem, R Thompson, G Satterfield, C Masse, G Mercer, B Neuhaus, I Cassias, E Chang, Y Lin, S Macmahon, J Wong, K Zhang, RE Smith. Collaborative Validation of the QuEChERS Procedure for the Determination of Pesticides in Food by LC-MS/MS. J Agric Food Chem. 2011 22;59(12):6383–411. doi: 10.1021/jf201618q.
- M Jemal. High-throughput quantitative bioanalysis by LC-MS/MS. Biomed. 2000 14(6):422–9. doi: 10.1002/1099-0801.
- RN Xu, L Fan, MJ Rieser, TA El-Shourbagy. Recent Advances in High-Throughput Quantitative Bioanalysis by LC-MS/MS. J Pharm Biomed Anal. 2007 28;44(2):342–55. doi: 10.1016.
- S Krishnaswami, H Möllmann, H Derendorf, G Hochhaus. A Sensitive LC-MS/MS Method for the Quantification of Fluticasone Propionate in Human Plasma. J Pharm Biomed Anal. 2000;22(1):123-9. doi: 10.1016/s0731–7085.
- BJ Molloy, A King, LG Mullin, LA Gethings, R Riley, RS Plumb, ID Wilson. Rapid Determination of the Pharmacokinetics and Metabolic Fate of Gefitinib in the Mouse Using a Combination of UPLC-MS/MS, UPLC-QToF/MS, and ion mobility (IM)-enabled UPLC-QToF/MS. Xenobiotica. 2021;51(4):434–446. doi: 10.1080/00498254.2020.
- IA Blair. Analysis of Endogenous Glutathione-Adducts and Their Metabolites. Biomed Chromatogr. 2010;24(1):29–38. doi: 10.1002/bmc.1374.
- Graichen ME, Neptun DA, Dent JG, Popp JA, and Leonard TB (1985) Effects of Methapyrilene on Rat Hepatic Xenobiotic Metabolizing Enzymes and Liver Morphology. Fundam Appl Toxicol 5:165–174.
- Geenen S, Guallar-Hoyas C, Michopoulos F, Kenna JG, Kolaja KL, Westerhoff HV, Thomas P, Wilson ID. HPLC-MS/MS Methods for the Quantitative Analysis of 5-oxoproline (pyroglutamate) inRrat Plasma and Hepatic Cell Line Culture Medium. J Pharm Biomed Anal. 2011 1;56(3):655–63. doi: 10.1016/j.jpba.2011.06.001.
720008016, September 2023