Fast and Robust LC-UV-MS Based Peptide Mapping Using RapiZyme™ Trypsin and IonHance™ DFA
Abstract
Laborious and time-consuming peptide mapping protocols are made faster and more robust by using a novel RapiZyme Trypsin and IonHance difluoroacetic acid (DFA). High activity and enhanced autolysis resistance enabled the RapiZyme Trypsin to be used at higher amounts leading to reduced digestion time (~1 hour) in contrast to the traditional enzyme-to-protein ratios and long digestion time (≥3 hours) employed in protein sequencing methodologies. The use of IonHance DFA as a mobile phase additive in the peptide mapping workflows made high-quality mass spectral (MS) and UV data acquisition possible without adduct-induced interferences. Using Waters MassPREP™ peptide standard mixture and NISTmAb as protein digest standards, we demonstrate the direct alignment of high-quality UV and MS data while maintaining the high signal intensity of sequence-informative fragment ions in the MSe-based fragmentation of adduct-free target peptide molecular ions. Thus, after annotating peptide peaks to specific retention times in the initial experiments, UV only runs can be considered for subsequent quality control (QC) analyses. Such improved methodologies are expected to accelerate the testing of pharmaceutical proteins as a result of the ease and efficiency with which high quality can now be obtained.
Benefits
- The use of RapiZyme trypsin enables 1:5 enzyme-to-protein ratio digestions, thereby reducing digestion times
- Total sample preparation time is under 3 hours, significantly less than many conventional peptide mapping workflows
- Versus formic acid, IonHance DFA results in enhanced UV sensitivity in a sequence-independent fashion allowing the monitoring of low-abundant peptides
- Ion pairing properties of IonHance DFA further improves the reproducibility of peptide retention times while maintaining a sensitive response without the associated adducts
Introduction
Peptide mapping is a technique used in proteomics & biopharmaceutical characterization to identify and characterize proteins. A single chain of protein is also referred to as a polypeptide. A polypeptide is distinguished by its sequence of amino acids connected through the peptide bonds. Proteomic studies involve breaking down proteins into smaller peptides through the cleavage of peptide bonds by a protease such as trypsin, followed by their separation and mass analysis by liquid chromatography-mass spectrometry (LC-MS). The resulting peptide map can be used to identify the protein and determine its primary structure. Robust peptide mapping is necessary for several reasons. First, it can be used to confirm the identity of a protein, which is important for quality control and validation purposes. Second, peptide mapping can be used to determine the primary structure of a protein, which can be essential to early research programs. Finally, peptide mapping can detect post-translational modifications in the amino acid sequence, which can affect protein function and stability.
This application note focuses on the use of RapiZyme Trypsin and IonHance DFA for peptide mapping and discusses the unique advantages of this combination to facilitate a quick peptide mapping workflow, simultaneous optimization of UV and MS responses, and adduct-free mass spectra.
Experimental
Sample Preparation
NIST mAb samples [10 µL at 10 µg/µL] were denatured with 90 µL of 6 M guanidine hydrochloride (GuHCl) [5.4 M final concentration] solution and reduced with 5 mM dithiothreitol (DTT) [2 µL of 250 mM] for 30 minutes at ambient conditions. This was followed by alkylation with 10 mM iodoacetamide (IAM) [3 µL of 350 mM] for another 30 minutes at ambient conditions.
Desalt Protocol
A digestion buffer consisting of 10 mM calcium chloride (CaCl2) in 0.1 M tris(hydroxymethyl)aminomethane hydrochloride (Tris-HCl) was first prepared from Waters Tris CaCl2 Buffer Salts pH 7.5, 4/pk (p/n: 186010111). A gravity flow size-exclusion chromatography (SEC)-based desalt cartridge was conditioned by loading 400 µL of the digestion buffer three times and discarding the eluent each time. 100 µL of the denatured NIST mAb sample was then loaded onto the column and flowthrough discarded. The column was washed with 100 µL of digestion buffer, discarding the flowthrough. After ensuring that the cartridges were free of residual drops, the sample collection container was placed under the cartridge, and the desalted protein was collected with 300 µL of the digestion buffer.
Protein Estimation
To ensure that the correct amount of the denatured and alkylated NIST mAb was used for the digestion, protein estimation was performed after the desalting step. Precise concentration readings were obtained by A280 measurements using a UV-Vis plate reader. Alternatively, protein estimation can also be achieved through UV-Vis measurements of the droplets. This optional step ensures the use of the correct protein amount for digestion so that the enzyme-to-protein ratio values are consistently maintained for reproducible signal behavior across multiple digests.
Protein Digestion
20 µg (in 200 µL) of the denatured, reduced, and alkylated NIST mAb was taken in a 300 µL PCR (polypropylene) tube, and 4 µg (in 4 µL) of RapiZyme Trypsin (p/n: 186010108) was added to perform the digestion at a 1:5 (enzyme: protein) ratio. This solution was incubated at 37 °C for 1 hour on a revolving PCR block @ 300 RPM. After digestion, the reaction was quenched by acidifying the samples with 20 µL of 1% formic acid (0.1% final concentration). The samples were vortexed and quantitatively transferred to Qsert vials for LC-MS analyses.
LC Conditions
|
LC system: |
ACQUITY™ UPLC™ I-Class PLUS |
|
Detection: |
ACQUITY TUV/PDA |
|
Wavelength: |
219 nm |
|
Vials: |
Clear Glass 12 x 32 mm Screw Neck Qsert Vial, 300 µL Volume (p/n: 186002804) |
|
Column(s): |
ACQUITY Premier Peptide CSH™ C18 Column, 130 Å, 1.7 µm, 2.1 x 150 mm (p/n: 186009489) |
|
Column temp.: |
65 °C |
|
Sample temp.: |
6 °C |
|
Injection volume: |
50 µL |
|
Flow rate: |
0.25 mL/min |
|
Mobile phase A: |
0.1% (v/v) IonHance DFA (p/n: 186009201) in LCMS-grade Water |
|
Mobile phase B: |
0.07% (v/v) IonHance DFA in LCMS grade Acetonitrile |
|
Gradient: |
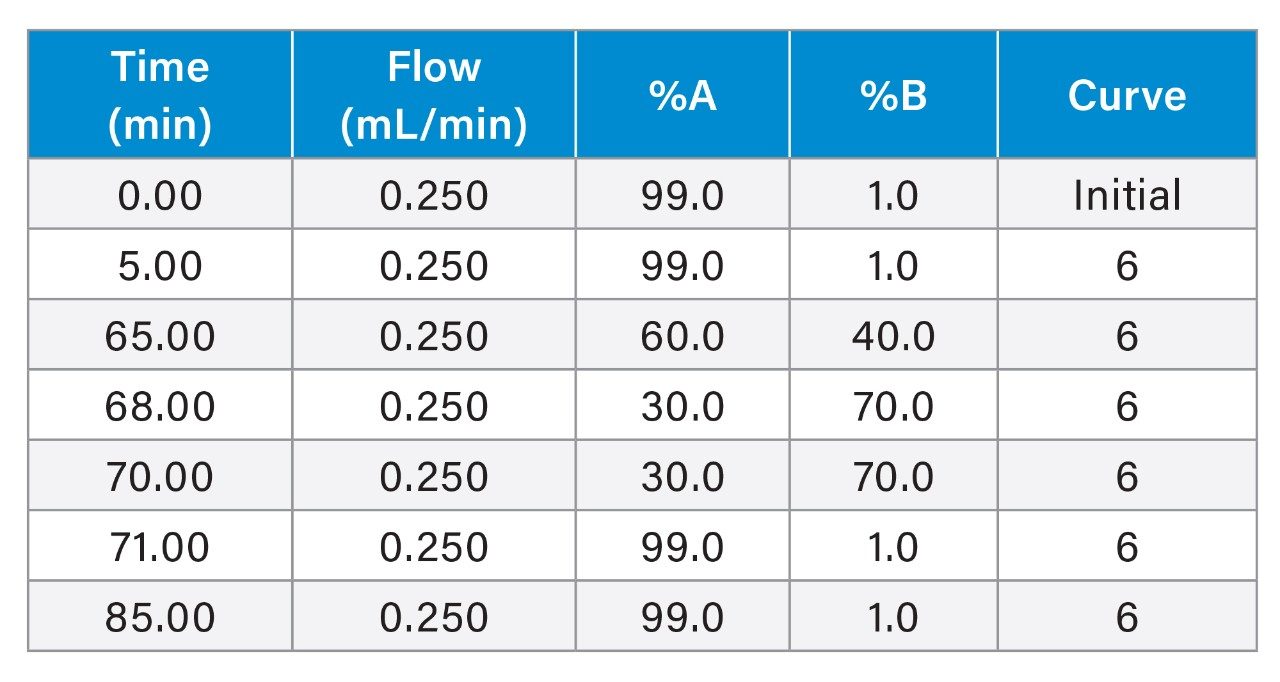
Please refer the table below. |
Gradient Table
MS Conditions
|
MS system: |
ACQUITY RDa |
|
Ionization mode: |
Full scan with fragmentation |
|
Acquisition range: |
Low (50–2000 m/z) |
|
Capillary voltage: |
1.20 kV |
|
Cone voltage: |
20 V |
|
Fragmentation cone voltage: |
60 V to 120 V |
|
Polarity: |
Positive |
|
Scan rate: |
2 Hz |
|
Desolvation temp.: |
350 °C |
Data Management
|
Chromatography software: |
UNIFI™ v 3.0.0.15, Empower™ 3 (For UV only analyses) |
|
MS software: |
UNIFI v 3.0.0.15 |
|
Informatics: |
UNIFI v 3.0.0.15 |
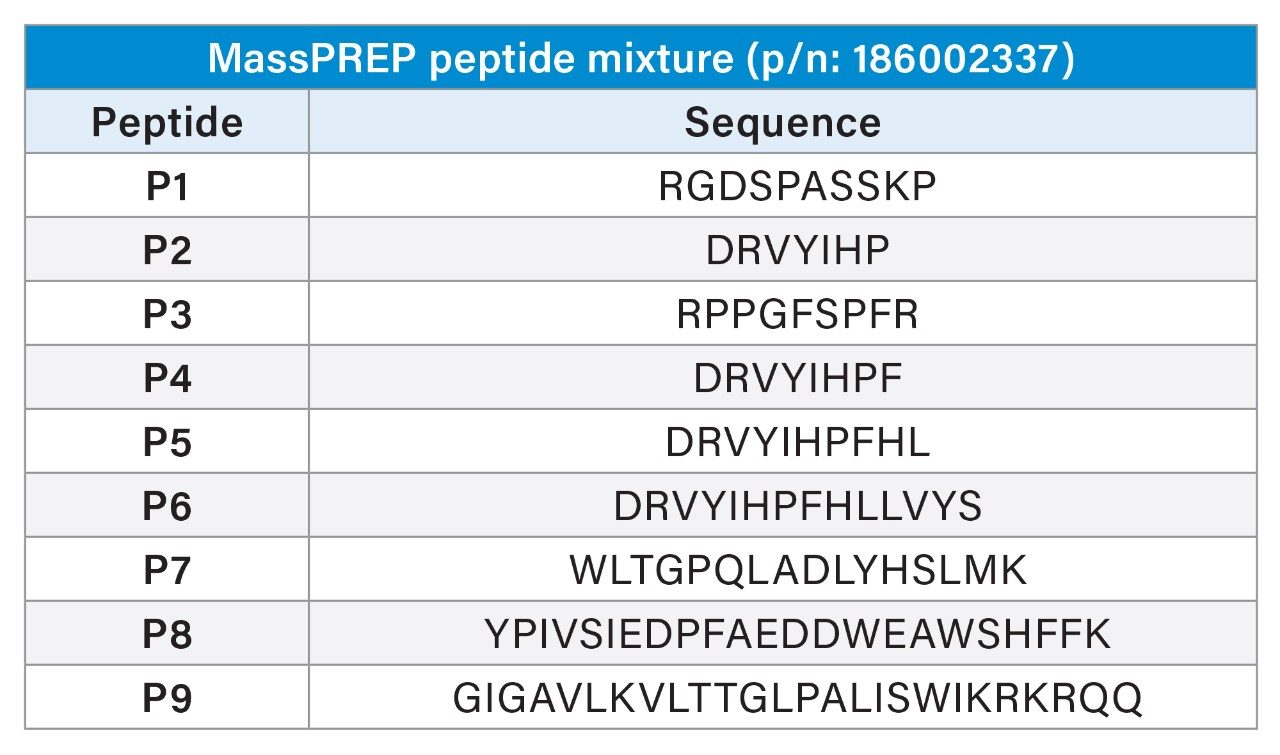 Peptide sequences of the nine peptides in the MassPREP Peptide Mixture
Peptide sequences of the nine peptides in the MassPREP Peptide Mixture
Results and Discussion
The current study intended to demonstrate the beneficial effects of RapiZyme Trypsin and IonHance DFA for use in quick and robust LC-UV-MS peptide mapping workflows. Using NIST mAb as an example biopharmaceutical protein of interest, we evaluated the efficacy of RapiZyme Trypsin at a 1:5 enzyme-protein ratio and 1 hour digestion time. The resulting peptides were subsequently subjected to LC-UV-MS analysis using IonHance DFA as a mobile phase additive in liquid chromatography.
This analysis revealed 100% amino acid sequence coverage of both the heavy and light chains (Figure 1). Like is expected for a formic acid mobile phase, the DFA modified mobile phases did not yield any ion pairing adducts (Figure 2) nor were there seen to be any adverse effects on the overall quality of the mass spectral data, including the acquisition of sequence-informative product ions. Further, the UV chromatograms exhibited enhanced UV signals with minimal baseline drift. The reproducibility of the protocol was demonstrated by the consistent UV and BPI chromatograms of separate, independently prepared protein digests (Figure 3–4).
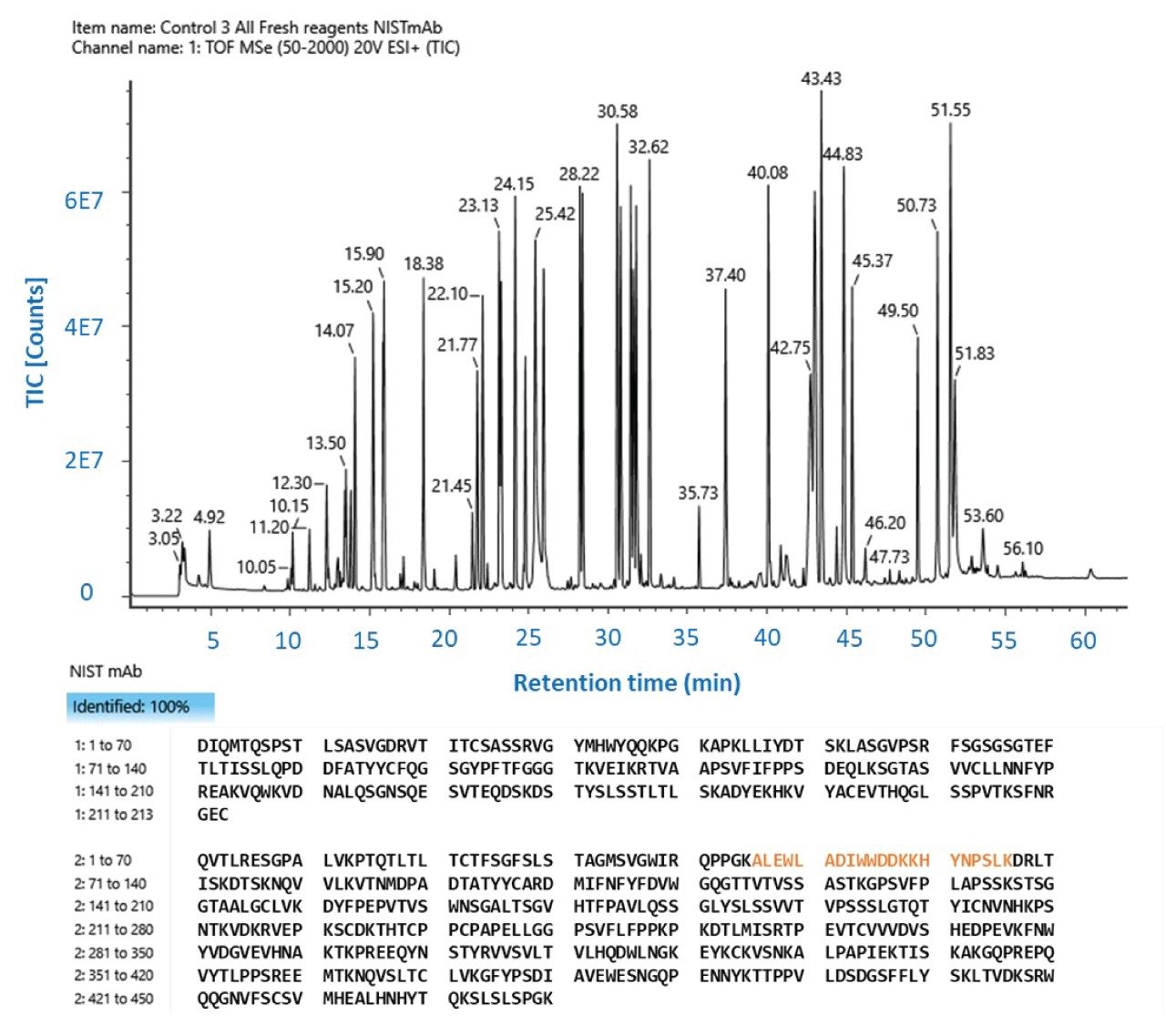 Figure 1. Representative total ion chromatogram (TIC) chromatogram and the observed sequence coverage of the NISTmAb sequence are shown. About 5 µg of protein digest was subjected to LC-UV-MS analysis using a BioAccord LC-MS system as described in the methods. The MSe data was analyzed by the UNIFI-based peptide mapping data analysis method to identify the high-confidence amino acid sequences of tryptic peptides and compute the sequence coverage.
Figure 1. Representative total ion chromatogram (TIC) chromatogram and the observed sequence coverage of the NISTmAb sequence are shown. About 5 µg of protein digest was subjected to LC-UV-MS analysis using a BioAccord LC-MS system as described in the methods. The MSe data was analyzed by the UNIFI-based peptide mapping data analysis method to identify the high-confidence amino acid sequences of tryptic peptides and compute the sequence coverage.
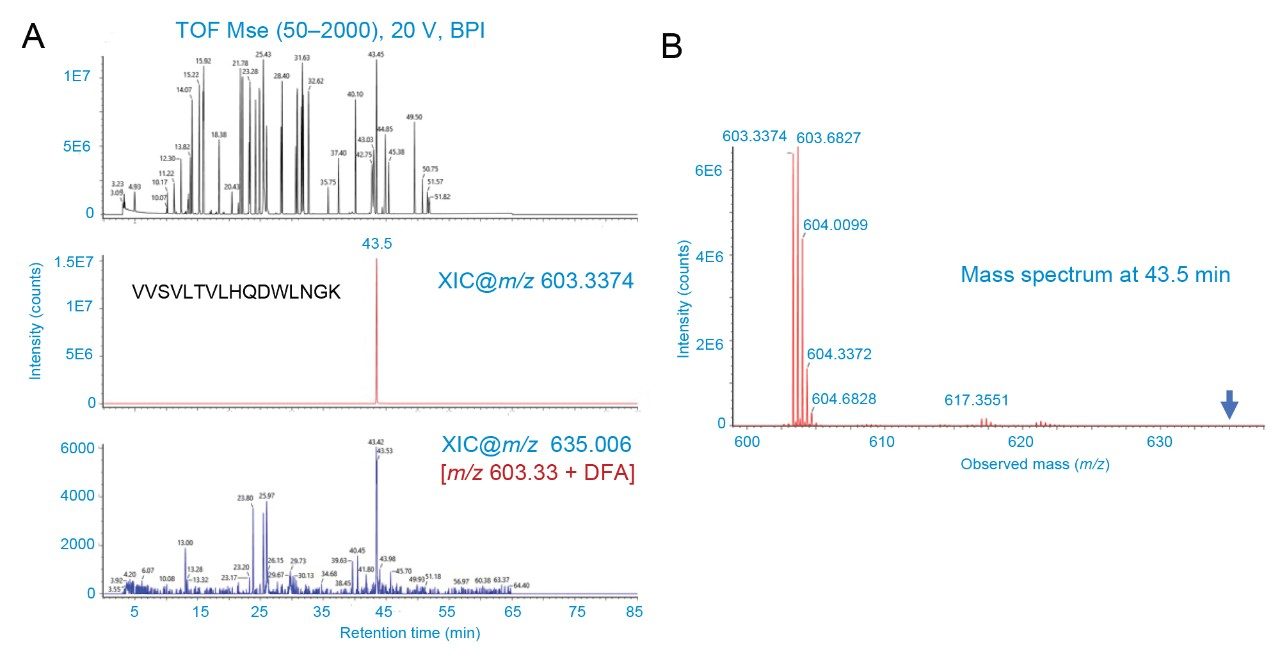 Figure 2. Analysis of LC-MS spectra for DFA adducts.
Figure 2. Analysis of LC-MS spectra for DFA adducts.(A) The top panel depicts the base peak ion chromatogram (BPI) of the NISTmAb protein digested with RapiZyme Trysin. The peptide peak at 43.5 min exhibited at highest ion abundance as seen in both BPI and extracted ion chromatogram (XIC) for m/z 603.3374 (middle panel) that corresponds to the peptide VVSVLTVLHQDWLNGK. If adducts are formed, this should be noticeable with this most abundant peptide signal. But the XIC for m/z 635.006 (bottom panel) corresponding to the DFA adduct (+95 Da after accounting for proton replacement) did not yield a clear peak as the signal was at baseline level (see the intensity for the bottom panel).
(B) The mass spectrum corresponding to 43–43.5 min did not show the presence of ions corresponding to the adducts (see the arrow).
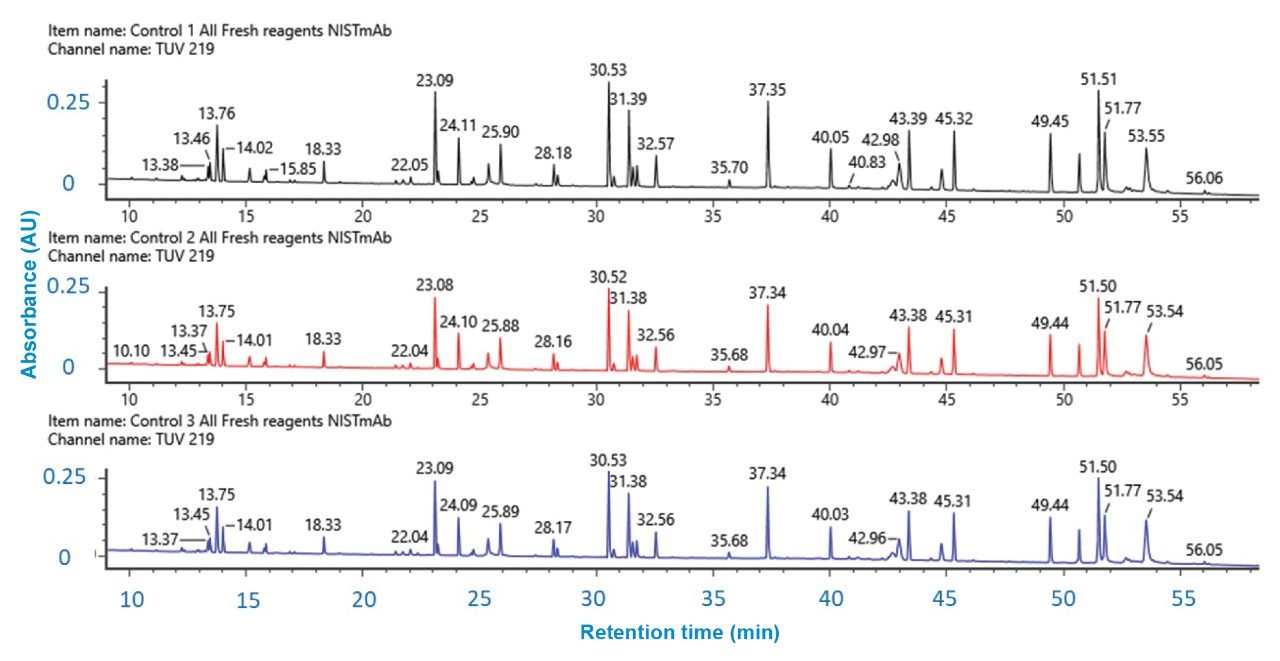 Figure 3. Reproducibility of UV chromatograms for three separate, independently prepared digest replicates of NISTmAb. The UV signal at 219 nm remained constant for all three digests indicating high reproducibility.
Figure 3. Reproducibility of UV chromatograms for three separate, independently prepared digest replicates of NISTmAb. The UV signal at 219 nm remained constant for all three digests indicating high reproducibility.
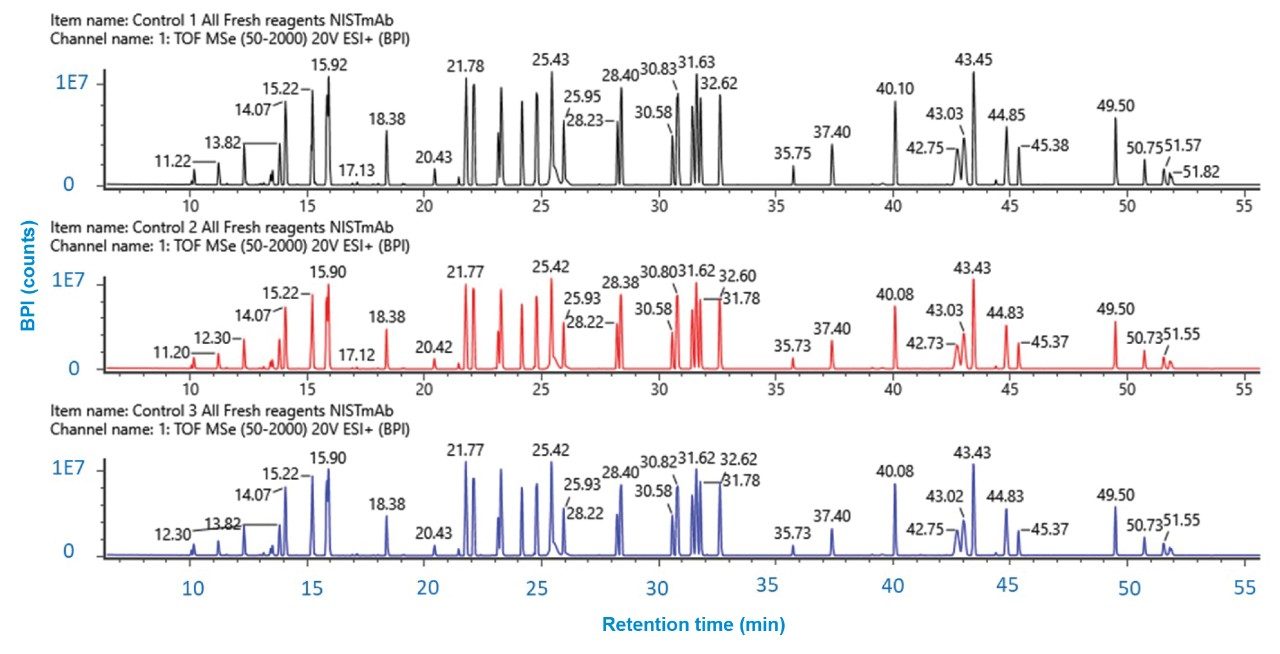 Figure 4. Reproducibility of BPI chromatograms for three separate, independently prepared digest replicates of NISTmAb. The UV signal at 219 nm remained constant for all three digests indicating high reproducibility.
Figure 4. Reproducibility of BPI chromatograms for three separate, independently prepared digest replicates of NISTmAb. The UV signal at 219 nm remained constant for all three digests indicating high reproducibility.
The first advantage of this protocol is the quick sample preparation process which is under 3 hours. This is partly due to the RapiZyme Trypsin’s ability to facilitate high enzyme:protein ratio digestions. The second advantage is that chromatograms showed high UV signal intensities. This can be attributed to differences in the refractive index properties of DFA versus formic acid as well as the improved peak shapes afforded by DFA ion pairing. The third is IonHance DFA's role in enabling adduct-free mass spectra. Finally, the excellent reproducibility of the peptide mapping was demonstrated by comparing the UV and BPI chromatograms between independent sample preparations.
Why do we need quick peptide mapping workflows?
Quick peptide mapping workflows can provide timely results to laboratories and companies working to research, develop and release protein drugs. Longer digestion and sample preparation times could introduce or allow the formation of sample artifacts, adversely impacting the analyses quality. Quick peptide mapping workflows can also enable the automation of high-throughput analysis in large-scale studies, making the QC analyses faster and less error-prone. A quicker turn around of results means quicker decisions, and fewer artifacts could help ensure that the right results are reported in the first pass of project work.
Why use IonHance DFA to enhance UV?
Formic acid is usually preferred over trifluoroacetic acid (TFA) in most LC-MS-based peptide mapping analyses because of the sensitive MS response and because adduct-free mass spectra are obtained. However, some peptides are not easily detectable with formic acid-based mobile phase (an example shown in Figure 5) because it does not have the ion pairing characteristics of TFA to improve chromatographic retention.1 This was evident by the behavior of MassPREP Peptide Mixture (p/n: 186002337) when subjected to UV-based detection. Peptide 1 was not detectable under these conditions at any of the wavelengths (210–225 nm) tested. It is not clear whether this peptide was pushed into the void volume partially or completely, leading to a loss of signal.
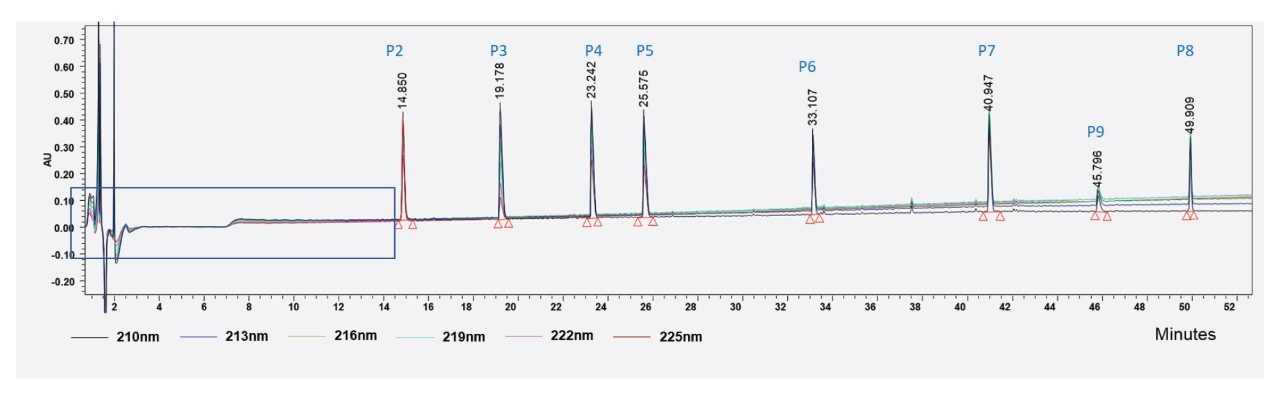 Figure 5. LC-UV-based detection of peptides from MassPREP Peptide Mixture (P1-P9) using formic acid-based mobile phase. The presence of P1 cannot be deciphered unambiguously from this profile (see the rectangular area on the chromatogram). Note the change in the elution pattern of P9 compared to P8.
Figure 5. LC-UV-based detection of peptides from MassPREP Peptide Mixture (P1-P9) using formic acid-based mobile phase. The presence of P1 cannot be deciphered unambiguously from this profile (see the rectangular area on the chromatogram). Note the change in the elution pattern of P9 compared to P8.
IonHance DFA, on the other hand, exhibits ion pairing characteristics (more like TFA); therefore, better retention and UV sensitivity may be expected in a peptide-independent fashion. Working with this insight, we sought to achieving the highest quality UV chromatograms possible. In order to identify the most suitable UV wavelength for peptide measurements when using IonHance DFA, the MassPREP Peptide Mixture was subjected to LC-UV analysis at detection wavelengths ranging from 210 to 225 nm. The nine peptides expected in the MassPrep peptide mixture could be detected in all situations despite the UV-baseline exhibiting wavelength-dependent drift (Figure 6). Although the maximum signal was noticed at 210 nm the baseline drifted downward, which may interfere with the quantitation. On the other hand, the baseline at 219 nm was comparatively steady, and the peptide signal did not suffer significant loss at this wavelength (Figure 7). Slightly lowering the concentration of DFA in mobile phase B resulted in further improvement and reproducible baseline behavior. Importantly, all the expected peptides, including peptide 1 were detected without any ambiguity. Thus, 219 nm was identified as the ideal wavelength to obtain a level baselines and peptide-independent UV signal acquisitions.
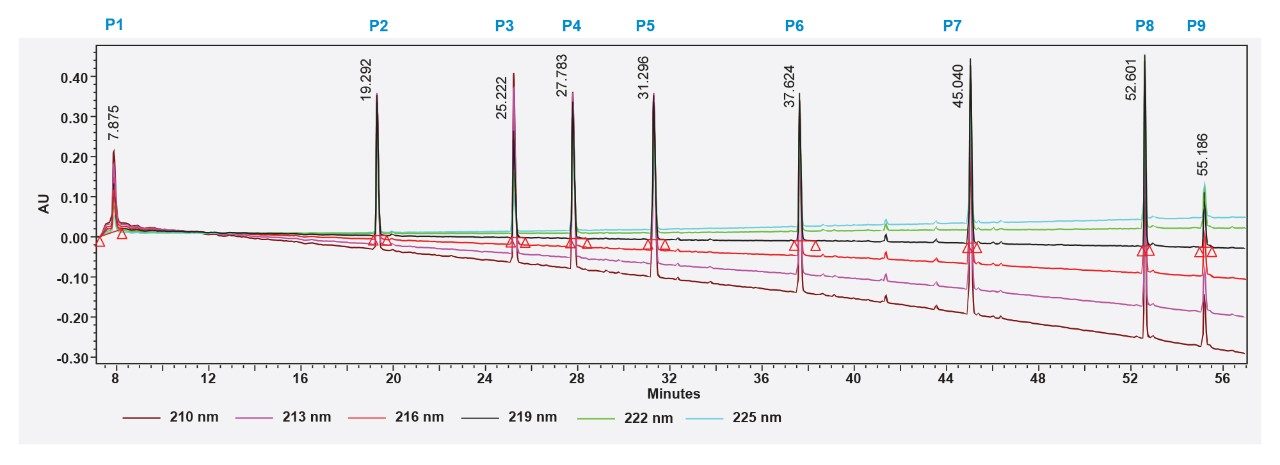 Figure 6. LC-UV-based detection of peptides from MassPREP Peptide Mixture (P1-P9) using IonHance DFA-based mobile phase. The presence of P1 can easily be deciphered unambiguously from this profile. Note the change in elution order and improved signal for P9 compared to the formic acid-based mobile phase.
Figure 6. LC-UV-based detection of peptides from MassPREP Peptide Mixture (P1-P9) using IonHance DFA-based mobile phase. The presence of P1 can easily be deciphered unambiguously from this profile. Note the change in elution order and improved signal for P9 compared to the formic acid-based mobile phase.
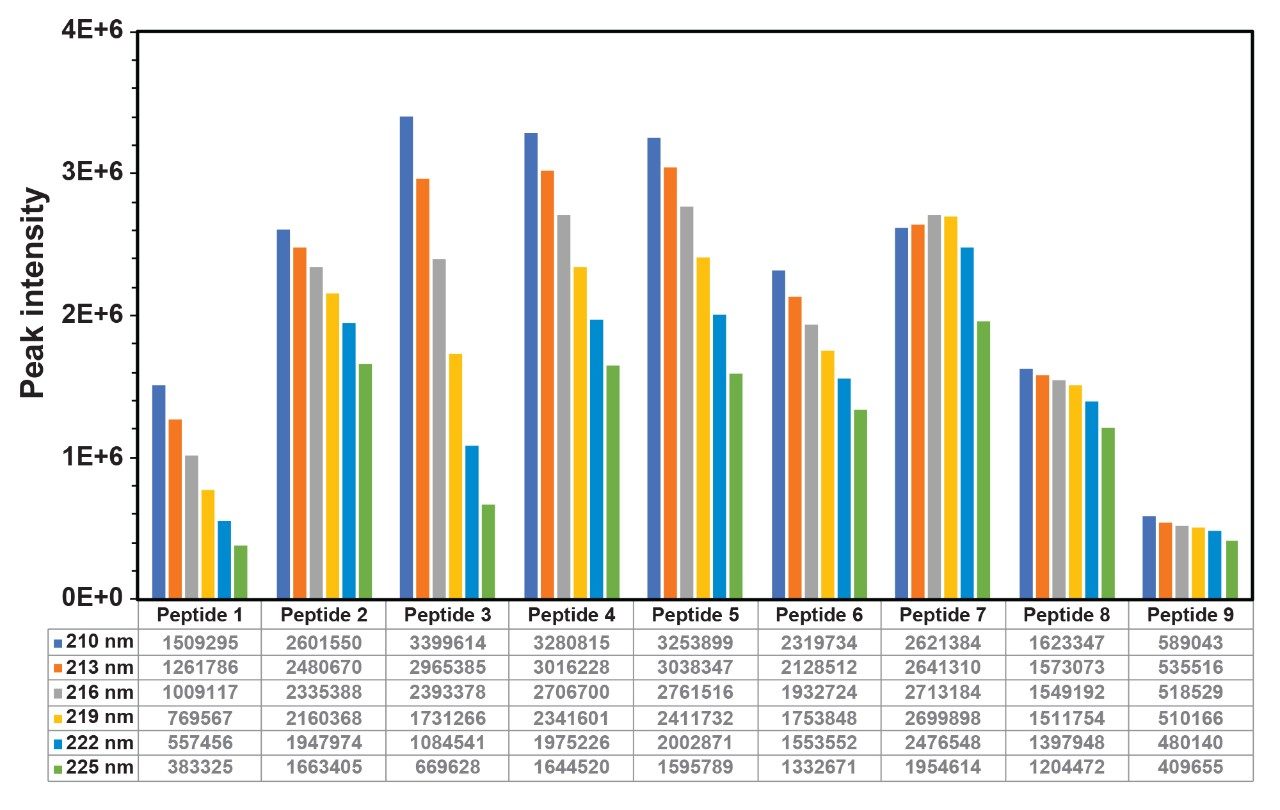 Figure 7. Peak area comparison of all the peptides detected by IonHance DFA-based mobile phase at UV lengths ranging from 210–225 nm.
Figure 7. Peak area comparison of all the peptides detected by IonHance DFA-based mobile phase at UV lengths ranging from 210–225 nm.
Strong, reliable ultraviolet (UV) signal can provide several advantages in peptide mapping.
- Improved sensitivity: Sequence-independent UV-based recognition allows efficient detection of low-abundance peptides.2
- Increased accuracy: Provides distinct and well-defined peaks in the chromatogram.3
- Enhanced resolution: Fewer co-elutions in the chromatogram, improving the analyses of complex peptide mixtures.2
- Enhanced reproducibility: Provides more consistent and reliable results.3
Also, in QC environments where users might not yet have LC-MS techniques, high-quality UV chromatograms can enable the analysts to obtain the required peptide mapping information without repeated MS data acquisition. This is because the initial LC-UV-MS analysis at the analytical level would help annotate the peptide peaks in the LC profiles, which can easily be transferred to QC situations without any intermediary data loss or ambiguity.
Conclusion
Peptide mapping workflows are typically complex and time-consuming. Hence, there is a need for simple, reproducible, and robust workflows that can facilitate reliable QC analyses and monitoring, as in the case of the biopharmaceutical industry. Sample preparation is a crucial step in these workflows and is usually time-consuming; however, RapiZyme Trypsin can enable users to reduce their sample preparation times, as demonstrated in this application note. Furthermore, the use of IonHance DFA as a mobile phase additive allows a robust LC-UV-MS peptide mapping experience through high-quality and reproducible data acquisition. This type of method show promise for becoming a platform method and enabling the timely assessment of protein therapeutical products in highly regulated environments.
References
- J. M. Nguyen, J. Smith, S. Rzewuski, C. Legido-Quigley, and M. A. Lauber, ‘High Sensitivity LC-MS Profiling of Antibody-Drug Conjugates With Difluoroacetic Acid Ion Pairing’, MAbs, vol. 11, no. 8, pp. 1358–1366, Nov. 2019, doi: 10.1080/19420862.2019.1658492/SUPPL_FILE/KMAB_A_1658492_SM8763.DOCX.
- M. L. Gross, G. Chen, and B. N. Pramanik, ‘Protein and Peptide Mass Spectrometry in Drug Discovery’, p. 464, 2012.
- T. Mouchahoir and J. E. Schiel, ‘Development of an LC-MS/MS Peptide Mapping Protocol for the NISTmAb’, Anal Bioanal Chem, vol. 410, no. 8, pp. 2111–2126, Mar. 2018, doi: 10.1007/S00216-018-0848-6/FIGURES/9.
720007864, February 2023