多重反射飛行時間型 MS および UPLC™ を用いた代謝物構造推定における ppb(10 億分の 1)単位の質量精度の影響の検討(パート B)
要約
SELECT SERIES™ 多重反射飛行時間(MRT)質量分析計を使用して、複数の医薬品および対応する代謝物の構造推定について、24 時間にわたるネガティブイオンエレクトロスプレー(ES-)モードでの LC-MS 精密質量測定の性能を評価しました。1813 種類の代謝物の検出において、RMS 誤差は 761 ppb であり、MSE データインディペンデント分析(DIA)のフラグメントイオンで ppb 単位の質量精度が得られています。
プリカーサーイオンおよびフラグメントイオンについて ppb 単位の精密質量測定値がルーチンに得られ、厳しい検出後データ解析の許容誤差を用いて、代謝物の構造推定が行われました。ppb 単位の質量精度により、尿スクリーニング分析で検出された低分子代謝物分子種の構造推定の信頼性が向上します。
UPLC-MSE ES- ノンターゲット DIA アッセイは、取り込み速度 10 Hz、システム質量分解能 200,000 FWHM 超で行われてきました。高質量分解能での妥協のない取り込みデューティサイクルにより、最適なクロマトグラフィーピークの忠実度が高まり、ppb 単位の質量精度と合わせて、共溶出する異性体分子種が確実に観測できます。
200,000 FWHM を超えるシステム質量分解能で観測された全体的な ppb 単位の質量精度と微細同位体構造の特性により、患者の尿サンプル中に存在する 3 種類の医薬品の代謝物が容易に同定されました。ここでは、報告例のあまりない代謝物が検出されました。
アプリケーションのメリット
- 分析効率が向上
- プリカーサーイオンおよび DIA MSE フラグメントイオンのルーチンでの ppb 単位の質量精度
- 微細同位体構造により追加の基準が得られ、低分子医薬品およびその代謝物の構造推定の信頼性が向上
- 200,000 FWHM を超えるシステム質量分解能により、複雑な生体マトリックス中の分析種の検出が向上
- 妥協のない 10 Hz 取り込みデューティサイクルにより、クロマトグラフィーピークの忠実度を保持
はじめに
SELECT SERIES MRT(図 1)は、非常に先進的なハイブリッド四重極 MRT 質量分析計です。取り込み速度に関係なく、高質量分解能(200,000 FWHM)とルーチンの ppb 単位の質量精度の組み合わせが得られます。四重極飛行時間型質量分析計(Q-ToF)などの高質量分解能質量分析計(HRMS)は、対象成分が複雑なマトリックス中に存在する多くの研究領域(食品・環境分析、天然物プロファイリング、メタボロミクス、臨床、法中毒学、代謝物同定など)におけるスクリーニングツールとして、普及が進んでいます。
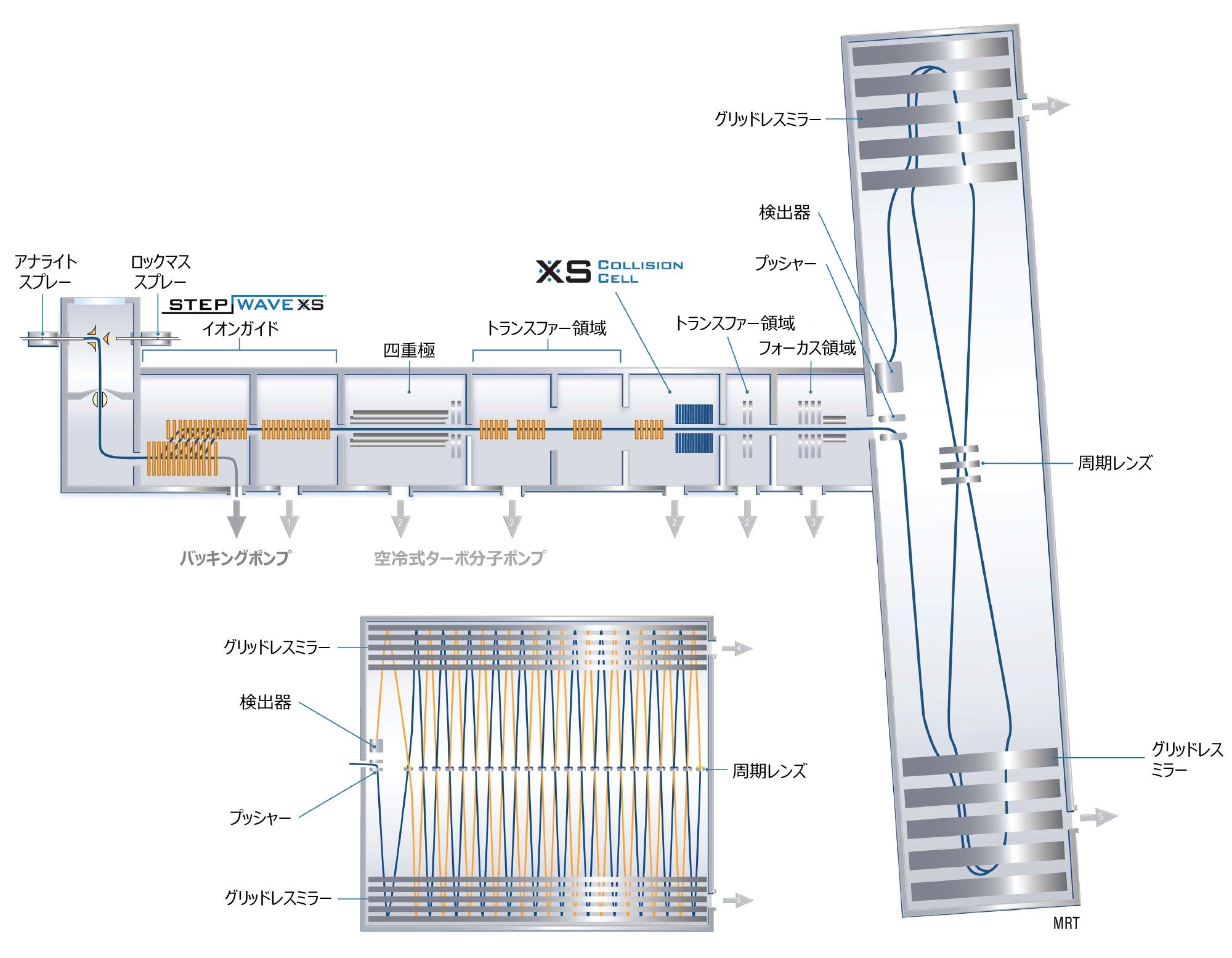 図 1. SELECT SERIES MRT 装置の概略図
図 1. SELECT SERIES MRT 装置の概略図
ターゲット取り込みメソッドを使用してスクリーニングできる化合物の数は通常限定されており、代表的な MS ライブラリーを生成するにはレファレンス標準試料が必要です。ターゲット分析戦略の場合は、未知化合物のマススペクトルデータは得られません。保持時間、分子式(精密質量)、および予想プロダクトイオンが取り入れられている質量分析ライブラリーデータベース(ライブラリーエントリー数が数十~数千)を、ノンターゲット取り込み戦略を使用する既知分析種のスクリーニングに使用します。続いて、適切なデータ解析フィルターを適用して、サンプル中の化合物の有無を判定し、何千もの分析種の検出によって生じる誤検出率を低減することができます1。 バイアスのないデータ取り込みは分析上の課題となりますが、サンプルの特性解析を完全に行い、同時に既知化合物と未知化合物を同定するにはこれが必要になります。
すべてのプリカーサーイオンとフラグメントイオンを取り込む DIA のようなバイアスのないデータ取り込み戦略を利用することで、サンプル全体の組成の代表的なプロファイルを容易に取得することができます。DIA MSE 取り込みは、MS/MS や DDA 法ほど選択的ではありませんが、200,000 FWHM を超える MRT システム分解能を使用することで、MSE の特異性が高まります2。高質量分解能により、複雑なマトリックス中の分析種のイオン選択性が向上することで、高い質量精度と検出能が得られ、結果として分析種の同定の信頼性が向上します。共溶出する同重体のマトリックス成分から、フラグメントイオンの質量を分離できます。その結果、保持時間でアライメントされたプリカーサー/フラグメントイオンを高い質量精度で観測でき、信頼性の高い CID スペクトルの解釈が容易に行えます。ppb 単位の高い質量精度のデータにより、生成される可能性のある元素組成の数が減り、その結果、分析種の同定において高い信頼性が得られます。MRT のルーチンでの ppb 単位の質量精度性能により、質の高い質量分析データが得られ、ノンターゲットスクリーニングワークフローで決定される分析種の元素組成に関して高い信頼性が容易に得られます。同位体微細構造および向上した質量精度特異性を併用して、代謝物構造推定などの低分子に関わる研究において、同定の信頼性を確実に改善することができます。
以前、ES+ 代謝物構造推定ワークフローを用いて、SELECT SERIES MRT に接続した Ultra Performance LC™(システム分解能 200,000 FWHM 超)を使用して、健常なボランティアのノンターゲット尿スクリーニングが行われました。[ウォーターズアプリケーションノート:720007896、2023 年 4 月] ナプロキセン、カルバマゼピン、アセトアミノフェンなどの医薬品およびそれらの代謝物が同定されました。高質量分解能の包括的なメリットを説明するため、ES- ノンターゲット尿スクリーニングを紹介します。
実験方法
サンプルの説明
10:1(H2O)希釈したヒト尿サンプル
カルバマゼピン投与量:2 × 200 mg 錠
アセトアミノフェン投与量:2 × 500 mg 錠
ナプロキセン投与量:1 × 500 mg 錠
サンプル採取のタイムポイント:薬物投与の 0、2、4、6 時間後
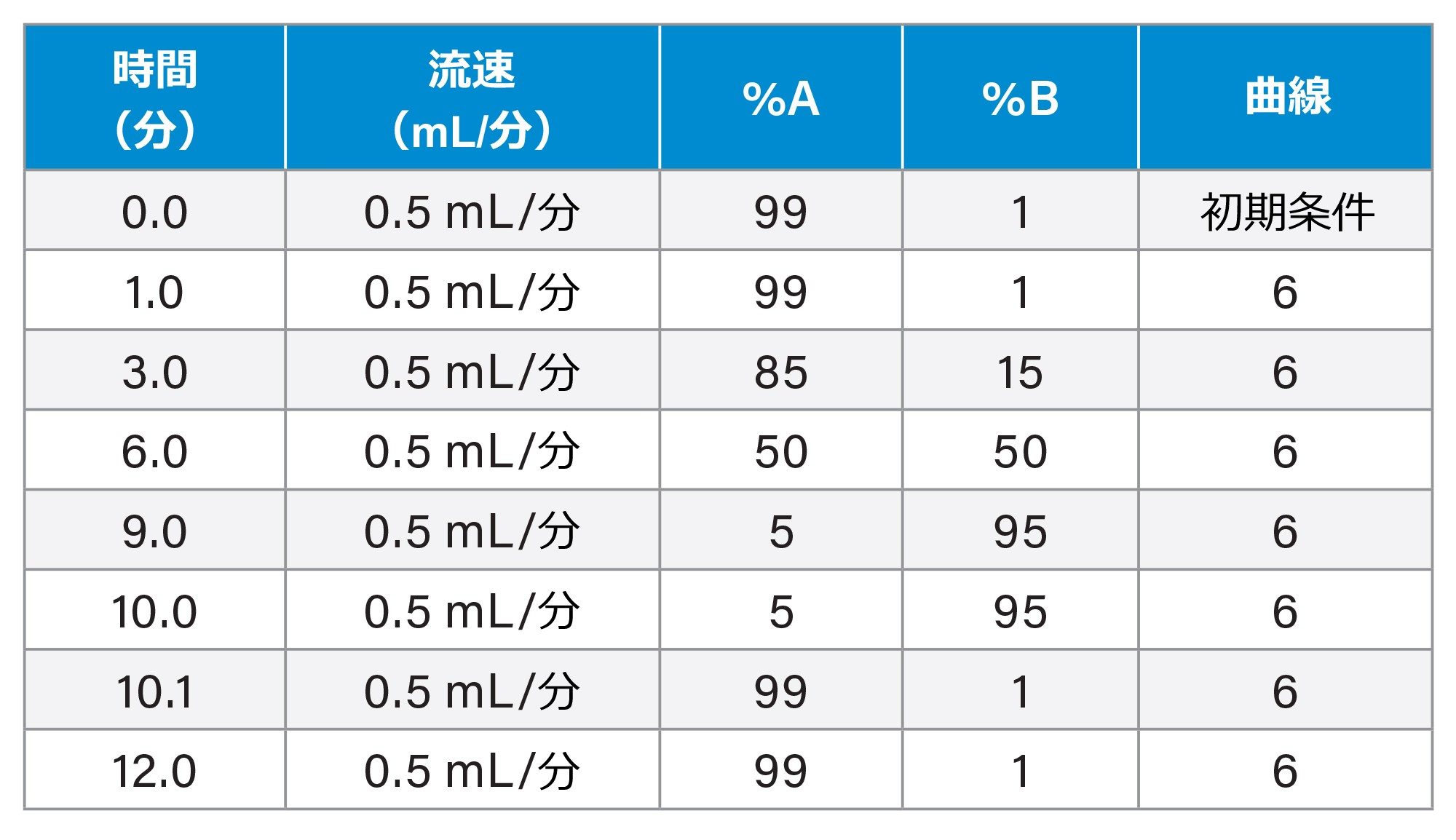
LC 条件
|
LC システム: |
Waters™ ACQUITY UPLC I-Class Premier クロマトグラフ |
|
カラム: |
ACQUITY UPLC HSS T3 C18(100 mm ×2.1 mm、1.8 μm)カラム |
|
カラム温度: |
40 ℃ |
|
サンプル温度: |
4 ℃ |
|
注入量: |
5 μL |
|
流速: |
0.5 mL/分 |
|
移動相 A: |
0.1% ギ酸水溶液(v/v) |
|
移動相 B: |
0.1% ギ酸アセトニトリル溶液(v/v) |
グラジエントテーブル
MS 条件
|
取り込み: |
ES- |
|
キャピラリー電圧: |
1 kV |
|
脱溶媒温度: |
500 ℃ |
|
イオン源温度: |
120 ℃ |
|
コーン電圧: |
20V |
|
MSE コリジョンエネルギーランプ: |
15 ~ 45 eV |
|
質量範囲: |
m/z 50 ~ 2400 |
|
取り込み速度: |
10 Hz |
|
取り込み/解析ソフトウェア: |
MassLynx v4.2 SCN1026 および waters_connect™ 3.1.0.243 |
結果および考察
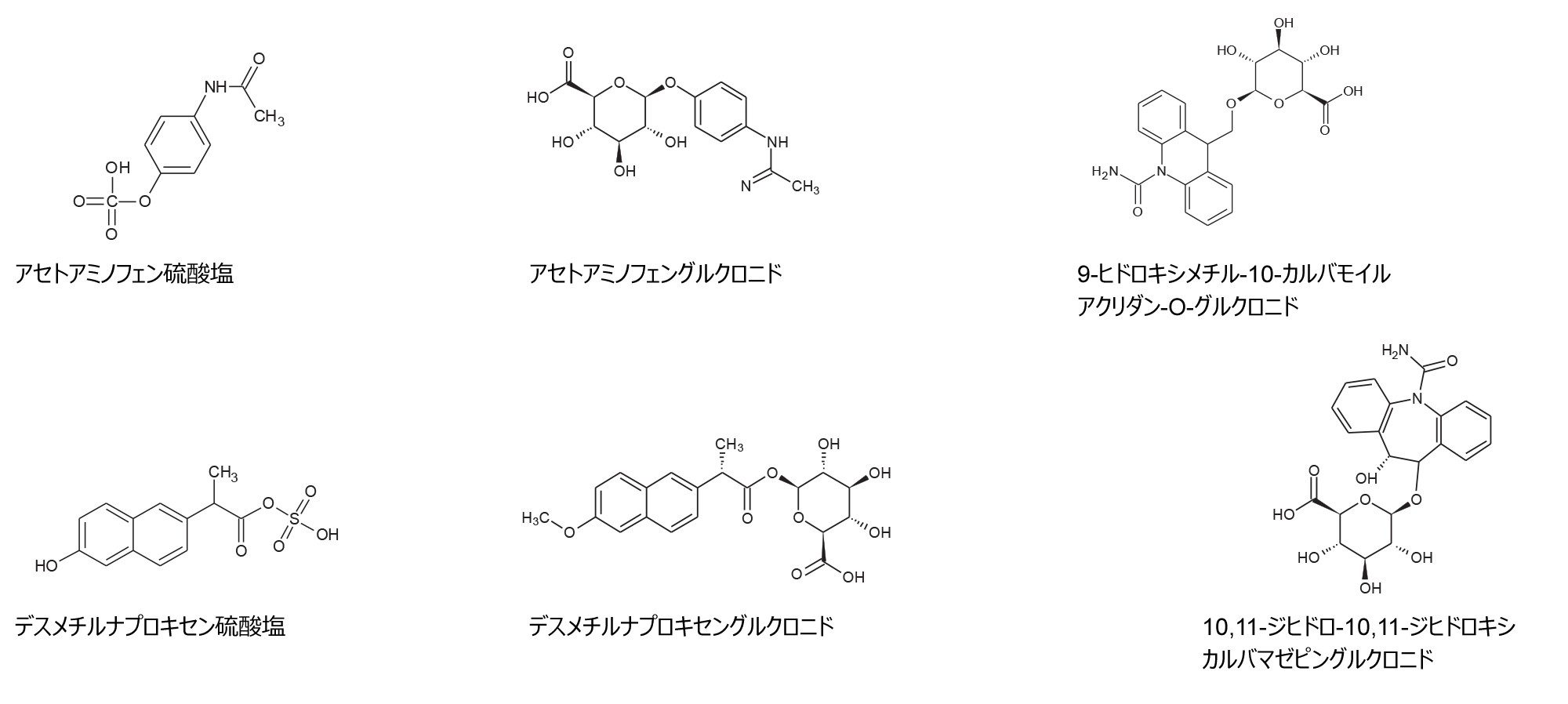 図 2. 代謝物構造推定ワークフローを使用した、医薬品アセトアミノフェン、カルバマゼピン、ナプロキセンの UPLC-MSE ES- ヒト尿スクリーニングで同定された代謝物の例
図 2. 代謝物構造推定ワークフローを使用した、医薬品アセトアミノフェン、カルバマゼピン、ナプロキセンの UPLC-MSE ES- ヒト尿スクリーニングで同定された代謝物の例
FDA MIST ガイドラインが 2008 年に発行されたことにより、代謝物の同定、定量および代謝物の対象範囲の改善において、製薬業界で採用される実験戦略に大きな変化が生じました4。 医薬品の探索および開発で提案された薬物候補において、代謝過程で反応性が高く毒性を有する可能性のある代謝物が生成されることが、継続的な課題となっています。薬物の毒性と関連しているアシル基やエステル基のグルクロン酸抱合体が形成した場合、薬物の使用中止に至っています5。 代謝物構造推定は、薬物分子の代謝経路を調査する医薬品開発プロセスの重要な部分です。生体内反応の同定を確実に行うための時間効率のよい臨床/非臨床試験が不可欠です。構造解析には、特異性の高い質量分析手法が必要です。今回、SELECT SERIES MRT と ACQUITY Ultra Performance LC の組み合わせを用いた、薬物代謝物の LC-MS 分析について説明します。医薬品および代謝物の構造推定のために、健常なボランティアの尿スクリーニングを、LC-MS および代謝物構造推定ワークフロー(分解能 > 200,000 FWHM)を用いて行いました。UPLC-MSE ES+ および UPLC-MSE ES- を行うことで、既知化合物と未知化合物の包括的分析への道が開けます。
健常なボランティアの LC-MS 尿スクリーニングを行ったところ、アセトアミノフェン(鎮痛薬)、ナプロキセン(非ステロイド性抗炎症薬)、カルバマゼピン(抗けいれん薬)などの医薬品がタイムポイントサンプル(投与後 2、4、6 時間)中に同定されました。図 2 に UPLC ES- を使用して同定された代謝物の例を示し、図 3 に、生物学的に複雑なヒト尿サンプルの分析のために、10 Hz で取得した MRT-LC-MS ES- クロマトグラフィーの複雑さの例を示します。カルバマゼピン-N-グルクロニドの抽出質量クロマトグラムを例にすると、3.6 秒のベースピーク幅にわたって 19 のデータポイントが取得されました。対応するコンティナムマススペクトルを図 3(II)に示します。ここでは、m/z 411 で 189,000 FWHM を超える質量分解能が得られました。データから、UPLC ES- DIA 分析を質量範囲全体にわたって行うことができ、この分析では低 m/z でデューティサイクルが損なわれることなく高質量分解能が保持され、クロマトグラフィーの忠実度が保持されることがわかります。
ここで説明する医薬品は、ES+ を使用した方がよくイオン化しています。一方、ES+ および ES- での代謝物スクリーニングアッセイが行われる ES- を使用すると、追加の代謝物の確認および探索が行えます。イオン化効率の改善に加えて、例えばフェーズ II グルクロン酸抱合体および硫酸抱合体の場合。重要な点として、ES+ および ES- の ppb 単位の質量精度がルーチンに得られることで、構造推定の信頼性が高まります。表 1 に示すように、24 時間にわたるヒト尿のタイムポイントサンプルの繰り返し分析において、アセトアミノフェン硫酸塩では 882 ppb、アセチルシステインアセトアミノフェンでは 740 ppb、アセトアミノフェングルクロニドでは 773 ppb の RMS 質量測定誤差が達成されました。全体として、表 1 に示す m/z 150 ~ m/z 427 の範囲の低分子で構成される代謝物での RMS 誤差は 761 ppb(検出数 N = 1813)でした。
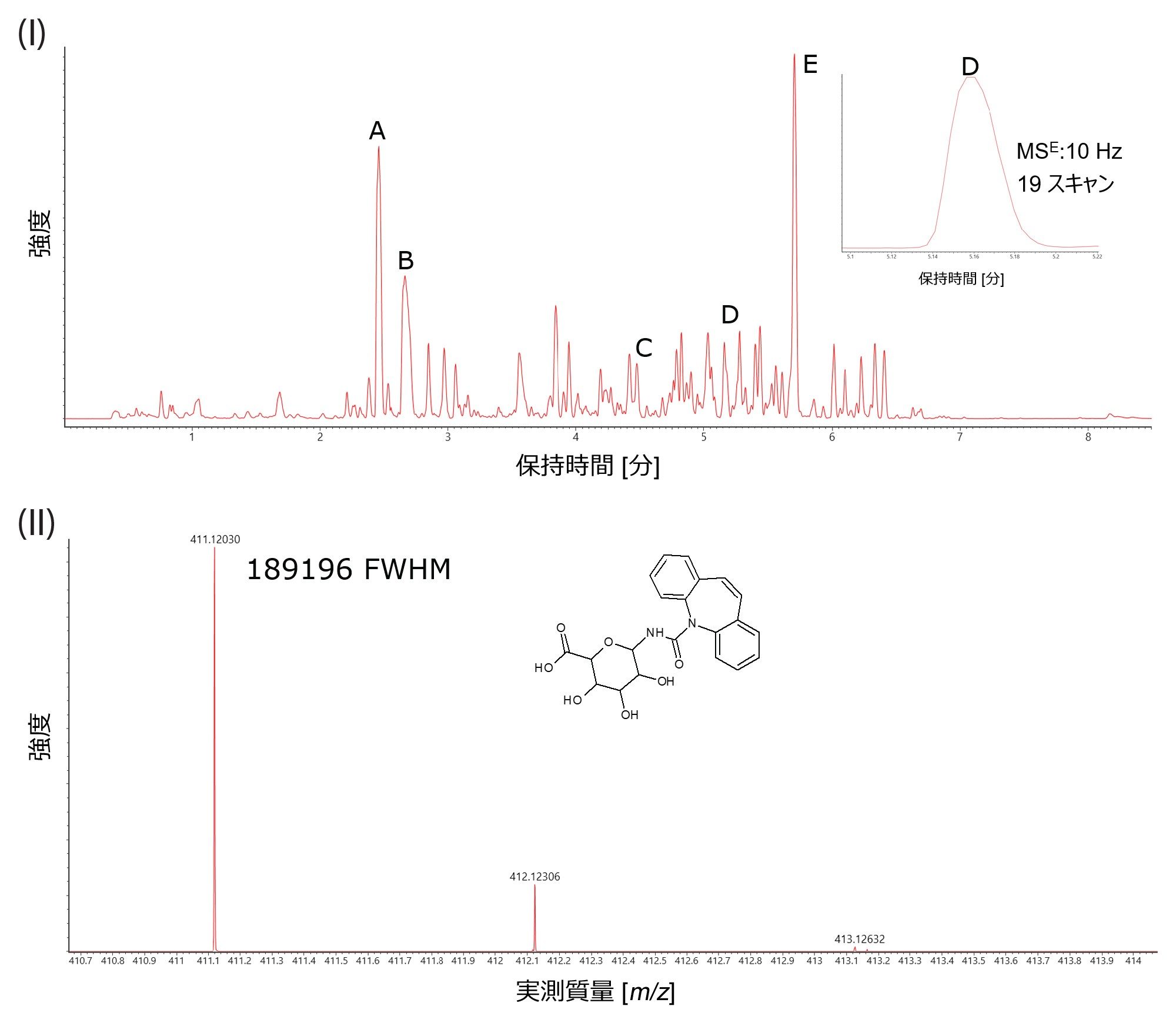 図 3. I)健常なボランティアの尿中に同定された医薬品(生体異物)および代謝物の分析で得られた MRT-LC-MS ES- 拡張ベースピークイオンクロマトグラム。A)アセトアミノフェングルクロニド、B)アセトアミノフェン硫酸塩、C)カルバマゼピン-O-グルクロニド、D)カルバマゼピン-N-グルクロニド、E)ナプロキセングルクロニド。挿入図は、カルバマゼピン-N-グルクロニドの質量クロマトグラムの拡大図。II) m/z 411 の [M-H]- カルバマゼピン-N-グルクロニドのマススペクトル。
図 3. I)健常なボランティアの尿中に同定された医薬品(生体異物)および代謝物の分析で得られた MRT-LC-MS ES- 拡張ベースピークイオンクロマトグラム。A)アセトアミノフェングルクロニド、B)アセトアミノフェン硫酸塩、C)カルバマゼピン-O-グルクロニド、D)カルバマゼピン-N-グルクロニド、E)ナプロキセングルクロニド。挿入図は、カルバマゼピン-N-グルクロニドの質量クロマトグラムの拡大図。II) m/z 411 の [M-H]- カルバマゼピン-N-グルクロニドのマススペクトル。
MSE を使用して DIA フラグメントイオンを生成し、マトリックス干渉から分離することにより、医薬品(生体異物)および代謝物の構造推定の信頼性がさらに向上します。図 4 に、10 Hz の UPLC MRT ES- を使用して、[アセトアミノフェン硫酸塩-H]- について得られたプリカーサーイオンおよびフラグメントイオンのスペクトルの例を示しています。測定誤差は -58 ppb(m/z 150) ~ 28 ppb(m/z 230)の範囲でした。フラグメントイオン m/z 80 では、1 ppm 以下の質量精度が観測され、10 Hz でそれぞれ 115,000 FWHM を超える質量分解能が得られています。通常のプリカーサーイオン/フラグメントイオンの ppb 単位の質量精度に加え、アセトアミノフェン硫酸塩の [M-H]- A+1 および A+2 同位体で示される特徴的な微細同位体構造により、追加の同定基準も得られます(図 4(II)参照)。
![10 Hz の UPLC MRT ES- を使用して、[アセトアミノフェン硫酸塩 -H]- について得られたプリカーサーイオンおよびフラグメントイオンのスペクトルII)10 Hz の UPLC MRT ES- を使用して得られた[アセトアミノフェン硫酸塩 -H]- の確認用詳細同位体構造](/content/dam/waters/ja/app-notes/2023/720007889/720007889en-f4.jpg.82.resize/img.jpg) 図 4 . I)10 Hz の UPLC MRT ES- を使用して、[アセトアミノフェン硫酸塩-H]- について得られたプリカーサーイオンおよびフラグメントイオンのスペクトル。II)10 Hz の UPLC MRT ES- を使用して得られた、[アセトアミノフェン硫酸塩-H]- の確認用詳細同位体構造。
図 4 . I)10 Hz の UPLC MRT ES- を使用して、[アセトアミノフェン硫酸塩-H]- について得られたプリカーサーイオンおよびフラグメントイオンのスペクトル。II)10 Hz の UPLC MRT ES- を使用して得られた、[アセトアミノフェン硫酸塩-H]- の確認用詳細同位体構造。
ナプロキセンの場合、ES+ でのクロマトグラフィーのピーク形状特性解析を使用して、1-β-O および 2,3,4-O 異性体であるアシルグルクロニド ES+ [M+Na]+ 分子種が、tr= 5.59 分、5.69/5.73 分(共溶出)および 5.86 分に観測されます。また、対応する [M-H]- 分子種が ppb 単位の質量精度でルーチンに検出されており、同定の信頼性が向上しています6,7。これらの分子種では、ES- でのイオン化効率が高いため、追加のナプロキセンアシルグルクロニド異性体が 5.95 分に同定され、RMS 質量測定誤差は 744 ppb でした。また、観測されたフラグメントイオンの ppb 単位の質量精度により、未知の異性体の同定の確実性が高まっています(図 5 参照)。
![10 Hz の UPLC MRT ES- を使用して、[ナプロキセングルクロニド-H]- について得られたプリカーサーイオンおよびフラグメントイオンのスペクトル](/content/dam/waters/ja/app-notes/2023/720007889/720007889en-f5.jpg.82.resize/img.jpg) 図 5. 10 Hz の UPLC MRT ES- を使用して、[ナプロキセングルクロニド-H]- について得られたプリカーサーイオンおよびフラグメントイオンのスペクトル
図 5. 10 Hz の UPLC MRT ES- を使用して、[ナプロキセングルクロニド-H]- について得られたプリカーサーイオンおよびフラグメントイオンのスペクトル
ES+ の [M+Na]+ に対応するデスメチルナプロキセングルクロニド [M-H]- 分子種が検出され(tr 4.83 分に観測)、構造推定は、プリカーサーイオンの RMS 質量測定誤差 950 ppb で確認されました。経口投与したナプロキセンおよびその代謝物の 95%(うち 85% がナプロキセングルクロニドまたは 6-O-デスメチルナプロキセングルクロニド)が尿中で回収されることが報告されています8。ナプロキセンには遊離の水酸基がないため、ナプロキセンの硫酸化は観測されません。一般的には報告されていませんが、6-O-デスメチルナプロキセンの硫酸化が観測されます。これが投与量の 11% を占め、tr 4.75 分にデスメチルナプロキセン硫酸塩([M-H]- m/z 295)が同定されました(質量精度 21 ppb 、図 6(I)を参照)9。代謝物構造推定が高い信頼性で確認され、プロダクトイオンである m/z 171 m/z(261 ppb)および m/z 143(28 ppb)が確認されました。さらに、特徴的な微細同位体構造により、追加の同定基準が得られます(図 6(III)を参照)10,11。図 6 に示されている DIA フラグメントスペクトルでは、m/z 331 にピークが観測されました。このピークにも、元素組成に硫黄が含まれる分子種であることを示す微細同位体構造(図 6(III)に表示)があります。ES- での代謝物構造推定ワークフローを使用して、カルバマゼピンのフェーズ II スルホン化生体内反応生成物が観測されることが確認されました。このような代謝物も一般には報告されていませんが、tr 4.75 分に、プリカーサーイオンである m/z 331(68 ppb)およびフラグメントイオン(m/z 251(216 ppb)/m/z 208(68 ppb))が観測されます12-16。取得されたプリカーサーイオンおよびフラグメントイオンのスペクトルの ppb 単位の質量精度がルーチンで再現性よく得られていることから、提案されたカルバマゼピンヒドロキシ硫酸塩代謝物の同定がさらに裏付けられます。図 6 から、ppm 以下の質量精度で DIA を使用すると、デスメチルナプロキセン硫酸塩分子種とカルバマゼピンヒドロキシ硫酸塩分子種のそれぞれのフラグメントイオンに相関があり、DIA 取り込み戦略を使用して非常に特異的なデータが得られたことがわかります。
最後に、観測された経時的レスポンスプロファイルは、真の生体内反応を示しています。この場合、観測された生体内反応分子種は、タイムコースのポイント T0 では検出されないか、分析した後のタイムコースポイントとは異なるレスポンスを示します。ES- アッセイの頑健性と質量精度を評価するために、溶媒ブランク試料の繰り返し注入(N = 3)、投与後 0、2、4、6 時間のタイムポイントで構成される繰り返し実験を行い、データセットの繰り返し分析を 24 時間にわたって継続して行いました。同定された医薬品(生体異物)であるアセトアミノフェン、カルバマゼピン-O-グルクロニド、ナプロキセンの代謝物トレンドプロットにより、図 7 では再現性が示されています。
![共溶出する I)[デスメチルナプロキセン硫酸塩 - H]- および II)[カルバマゼピン-O-硫酸 - H]- について得られたプリカーサーイオンおよびフラグメントイオンのスペクトルの ES- データインディペンデント分析。III)10 Hz の UPLC MRT ES- を使用して得られた(A)カルバマゼピン-O-硫酸- および(B)デスメチルナプロキセン硫酸塩の確認用 A+2 微細同位体構造](/content/dam/waters/ja/app-notes/2023/720007889/720007889en-f6.jpg.82.resize/img.jpg) 図 6. 共溶出する I)[デスメチルナプロキセン硫酸塩-H]- および II)[カルバマゼピン-O-硫酸-H]- について得られたプリカーサーイオンおよびフラグメントイオンのスペクトルの ES- データインディペンデント分析。III)10 Hz の UPLC MRT ES- を使用して得られた(A)カルバマゼピン-O-硫酸- および(B)デスメチルナプロキセン硫酸塩の確認用 A+2 微細同位体構造。
図 6. 共溶出する I)[デスメチルナプロキセン硫酸塩-H]- および II)[カルバマゼピン-O-硫酸-H]- について得られたプリカーサーイオンおよびフラグメントイオンのスペクトルの ES- データインディペンデント分析。III)10 Hz の UPLC MRT ES- を使用して得られた(A)カルバマゼピン-O-硫酸- および(B)デスメチルナプロキセン硫酸塩の確認用 A+2 微細同位体構造。
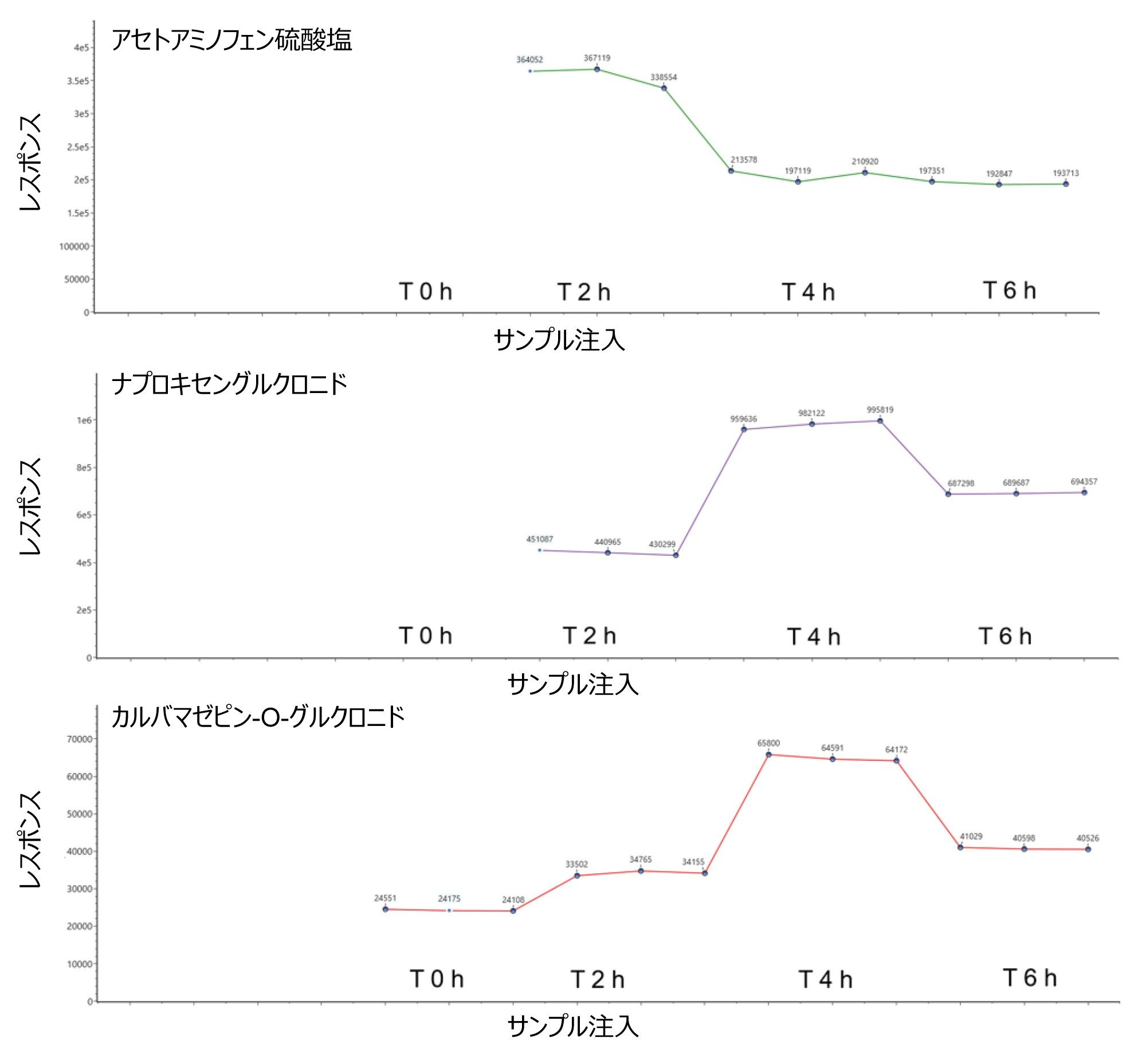 図 7. 10 Hz の UPLC MRT ES- および代謝物構造推定ワークフローを使用して得られた、ヒト尿サンプル中に同定された医薬品代謝物についての代謝レスポンストレンドプロット(投与後 0、2、4、6 時間のタイムポイント)
図 7. 10 Hz の UPLC MRT ES- および代謝物構造推定ワークフローを使用して得られた、ヒト尿サンプル中に同定された医薬品代謝物についての代謝レスポンストレンドプロット(投与後 0、2、4、6 時間のタイムポイント)
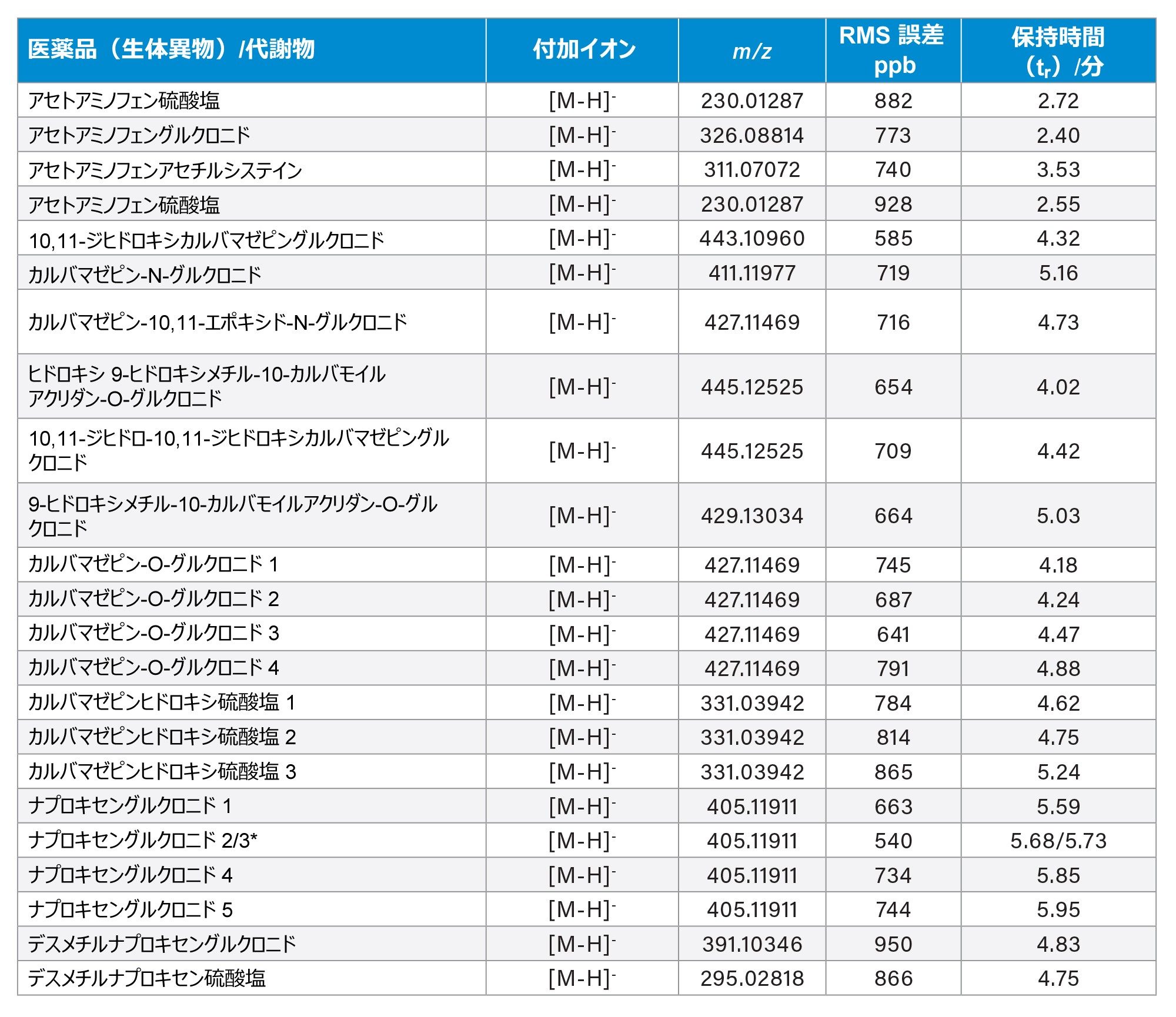 表 1. UPLC ES- を使用して 24 時間にわたって行った分析における、医薬品(生体異物)および代謝物検出のための RMS 精密質量測定値
表 1. UPLC ES- を使用して 24 時間にわたって行った分析における、医薬品(生体異物)および代謝物検出のための RMS 精密質量測定値
結論
LC-MS ネガティブイオンエレクトロスプレーを 10 Hz で使用する(200,000 FHWM を超えるシステム分解能を有する)ことにより、複雑な尿のノンターゲットスクリーニング分析において、プリカーサーイオンおよびフラグメントイオンについてルーチンに ppb 単位の質量精度が得られました。24 時間にわたって RMS 754 ppb レベルの質量精度が得られたことにより、ボランティアの尿サンプル中の、医薬品の組み合わせの主要代謝物および微量代謝物の確実な同定が可能になりました。全体的に見て、ここで示されている高分解能の ppb 単位の質量測定性能により、低分子の同定で決定される可能性のある元素組成の数が減ります。これは、既知化合物と未知化合物を取り扱う場合に大きなメリットになります。ナプロキセンの場合、5 種類の O-グルクロニド分子種が同定されました。すべてが完全に分離されていたわけではありませんが、10 Hz のデューティサイクルで異性体クロマトグラフィーの忠実度が保持されている 5 番目の分子種の同定を確実に行うことができました。プリカーサーイオン/フラグメントイオンの ppb 単位の質量精度と組み合わせることで、インフォマティクス代謝物ワークフローの割り当てから確信を得ることができました。この複雑な分析では、精密同位体構造が非常に有用であることがわかりました。取り込まれた特徴的な同位体分子種プロファイルを追加の同定基準として、理論構造と比較しました。これにより、スルホン酸ヒドロキシカルバマゼピンの生体内反応分析種と共溶出するフェーズ II 代謝物であるデスメチルナプロキセン硫酸塩の同定で示されているように、代謝物構造推定の信頼性が高まります。これらの構造推定は、データの質およびインフォマティクスソリューションのアウトプット、DIA プリカーサーイオンおよびフラグメントイオンの ppb 単位での質量測定の間の相互依存的相関を示しており、ノンターゲット分析取り込み戦略に高度の特異性が提供されます。質量分解能の向上による変革的な質量測定により、医薬品の探索および開発のプロセスにおいて、インフォマティクスのアウトプット、そしてさらには試験効率を向上させることができます。高分解能質量分析により、より厳格なインフォマティクスデータ解析フィルターの導入が容易になり、親薬物、代謝物のプリカーサー、予想フラグメントイオン、in-silico フラグメントイオンの同定の信頼性が高まります。
10 Hz の UPLC-MRT- MSE の ppb 単位の質量精度性能を、代謝物構造推定ワークフローとの関連で示しました。ここで、デューティーサイクルによって促進されるクロマトグラフィーの忠実度および ppb 単位の質量精度によって、多くの低分子アプリケーション分野における同定の信頼性が高まっています17-19。
参考文献
- Schymanski EL, Jeon J, Gulde R, Fenner K, Ruff M, Singer HP, Hollender J. Identifying Small Molecules via High Resolution Mass Spectrometry: Communicating Confidence.Environ.Sci.Technol. 2014, 48 (4), 2097–2098.
- Cooper-Shepherd DA, Wildgoose J, Kozlov B, Johnson WJ, Tyldesley-Worster R, Palmer ME, Hoyes JB, McCullagh M, Jones E, Tonge R, Marsden-Edwards E, Nixon P, Verenchikov A, Langridge JI.Novel Hybrid Quadrupole-Multireflecting Time-of-Flight Mass Spectrometry System.J Am Soc Mass Spectrom.2023 Feb 1;34(2):264–272.
- McCullagh M, Palmer M, Marsden-Edwards E, Cooper-Shepherd D and James Langridge, Exploring the Impact of Part Per Billion Mass Accuracy for Metabolite Identification using Multi Reflecting Time-Of-Flight MS with UPLC Part A. Waters appication note 720007896, April 2023.
- FDA, US."Guidance for industry: safety testing of drug metabolites." Center for Drug Evaluation and Research, Rockville, MD, USA (2008).
- Bradshaw PR, Athersuch TJ, Stachulski AV, Wilson ID.Acyl glucuronide reactivity in perspective,Drug Discovery Today, Volume 25, Issue 9, 2020,Pages 1639–1650.
- Mortensen RW, Corcoran O, Cornett C, Sidelmann UG, Troke J, Lindon JC, Nicholson JK, Hansen SH.LC-1H NMR used for determination of the elution order of S-naproxen glucuronide isomers in two isocratic reversed-phase LC-systems.J Pharm Biomed Anal.2001 Jan;24(3):477–85.
- Higton D , Wilson ID, Vissers JPC , Plumb RS.An automated method to study the rapid intramolecular transacylation of drug acyl glucuronides using Cyclic Ion Mobility Spectrometry-Mass Spectrometry Waters Corporation Poster: PSTR135089718.
- Falany CN, Ström P, Swedmark S. Sulphation of o-desmethylnaproxen and related compounds by human cytosolic sulfotransferases.Br J Clin Pharmacol.2005 Dec;60(6):632–40.
- Kiang CH, Lee C, Kushinsky S. Isolation and identification of 6- desmethylnaproxen sulfate as a new metabolite of naproxen in human plasma.Drug Metab Dispos 1989; 17: 43–8.
- Nagao T, Yukihira D, Fujimura Y, et al. Power of isotopic fine structure for unambiguous determination of metabolite elemental compositions: in silico evaluation and metabolomic application.Anal Chim Acta.2014;813:70–76.
- Ipsen A. Efficient calculation of exact fine structure isotope patterns via the multidimensional Fourier transform. Anal Chem.2014;86(11):5316–5322.
- Lertratanangkoon K and Horning M G. Metabolism of carbamazepine.Drug Metabolism and Disposition January 1982, 10 (1) 1–10.
- Bahlmann A, Brack W, Schneider RJ, Krauss M. Carbamazepine and its metabolites in wastewater: Analytical pitfalls and occurrence in Germany and Portugal.Water Res.2014 Jun 15;57:104–14.doi: 10.1016/j.watres.2014.03.022. Epub 2014 Mar 19.PMID: 24704908.
- Richter WJ, Kriemler P, Faigle JW.(1978).Newer Aspects of the Biotransformation of Carbamazepine: Structural Characterization of Highly Polar Metabolites.In: Frigerio, A. (eds) Recent Developments in Mass Spectrometry in Biochemistry and Medicine.Springer, Boston, MA.https://doi.org/10.1007/978-1-4613-3991-5_1
- Russell JL, Spiller HA, Baker DD.Markedly elevated carbamazepine-10,11-epoxide/carbamazepine ratio in a fatal carbamazepine ingestion, Case Rep.Med.2015 (2015), 369707.
- Kerr BM, Thummel KE.Wurden CJ, Klein SM, Kroetz DL, Gonzalez FJ, Levy R, Human liver carbamazepine metabolism: Role of CYP3A4 and CYP2C8 in 10,11-epoxide formation, Biochemical Pharmacology, Volume 47, Issue 11, 1994, Pages 1969–1979, ISSN 0006–2952.
- Batucan NSP, Tremblay LA, Northcott GL, Matthaei CD.Medicating the environment? A critical review on the risks of carbamazepine, diclofenac and ibuprofen to aquatic organisms, Environmental Advances, Volume 7,2022, 100164.
- Gago-Ferrero p, Bletsou AA, Damalas DE, Aalizadeh R, Alygizakis NA, Singer HP, Hollender J, Thomaidis NS.Wide-scope target screening of >2000 emerging contaminants in wastewater samples with UPLC-Q-ToF-HRMS/MS and smart evaluation of its performance through the validation of 195 selected representative analytes. Journal of Hazardous Materials, Volume 387, 2020, 121712.
- Carere M, Polesello S, Kase R, Gawlik BM.(2015).The Emerging Contaminants in the Context of the EU Water Framework Directive.In: Petrovic M, Sabater S, Elosegi A, Barceló, D. (eds) Emerging Contaminants in River Ecosystems.The Handbook of Environmental Chemistry, vol 46.Springer, Cham.
720007889JA、2023 年 4 月