High Resolution Mass Spectrometry Metabolic Profiling of Human Urine Using the SELECT SERIES™ MRT
Abstract
Metabolic profiling of complex biological matrices can help understand the impact of exogenous factors on human health and disease progression. The matrices utilized in these metabolomic studies contain many diverse analytes and therefore require high resolution instrumentation to deconvolve the many different compounds present. Here the SELECT SERIES MRT enabled the confident sub ppm annotation of unknown metabolites in human urine and leveraged high mass resolution to further elevate the confidence in elemental composition using resolved fine isotopic distributions.
Benefits
- Higher confidence identifications reduce burden on subsequent validation steps
- Ultra-high resolution and ppb mass accuracy, independent of acquisition rates, increase compatibility with a wider choice of methods
- Fine isotopic structure improves confidence in elemental composition calculations
- Efficient, guided informatics workflows facilitate data interpretation and results generation
Introduction
High resolution mass spectrometry has been widely used in metabolomic studies to provide valuable accurate mass measurements for unknown molecules detected in biological matrices. Identifying these unknown compounds from human studies, is vital in helping understand underlining biological mechanisms, markers of disease progression or the impact of exogenous factors on the body. Therefore, the more accurate and precises these measurements are, the greater the confidence in identifying the compound can be. Additionally, the high mass resolution of modern instruments can resolve finer isotopic patterns helping the determination of elemental compositions and confirm adduct information. These higher resolution instruments can also help distinguish between two co-eluting compounds of similar molecular weight, where lower resolution instruments would struggle. Whilst high-resolution mass spectrometry is powerful in its own right, the ability to couple these characteristics with liquid chromatography is highly beneficial in further increasing metabolome coverage. Researchers within the field use a variety of different workflow options to process their data but all follow the same principle of peak picking, normalization, statistical analysis, and eventual feature annotation.1 Here we describe the utilization of the SELECT SERIES MRT mass spectrometer in the analysis of human urine samples and its integration with Waters™ metabolomics workflow and third-party statistical tools.2 The unique capabilities of the SELECT SERIES MRT which include sub ppm mass accuracy and high mass resolution achievable at fast UPLC acquisition rates, helps provide greater confidence during significant feature annotation.
Experimental
Sample Description
Human urine samples from the National Institute of Standards and Technology (NIST) were purchased from MERCK and contained pooled urine from three groups of donors, smokers, non-smokers, and passive smokers. An aliquot of each pooled sample was diluted with deionized water at a ratio of 1:4 where the sample was vortex mixed and centrifuged at 13,000 rpm for 10 minutes.
LC Conditions
|
LC system: |
ACQUITY™ Premier UPLC™ |
|
Column: |
ACQUITY Premier HSS T3, 1.8 µm, 2.1 x 100 mm |
|
Column temperature: |
45 °C |
|
Sample temperature: |
4 °C |
|
Injection volume: |
5 µL |
|
Flow rate: |
0.6 mL/min |
|
Mobile phase A: |
Water w/ 0.1% formic acid |
|
Mobile phase B: |
Acetonitrile w/ 0.1% Formic acid |
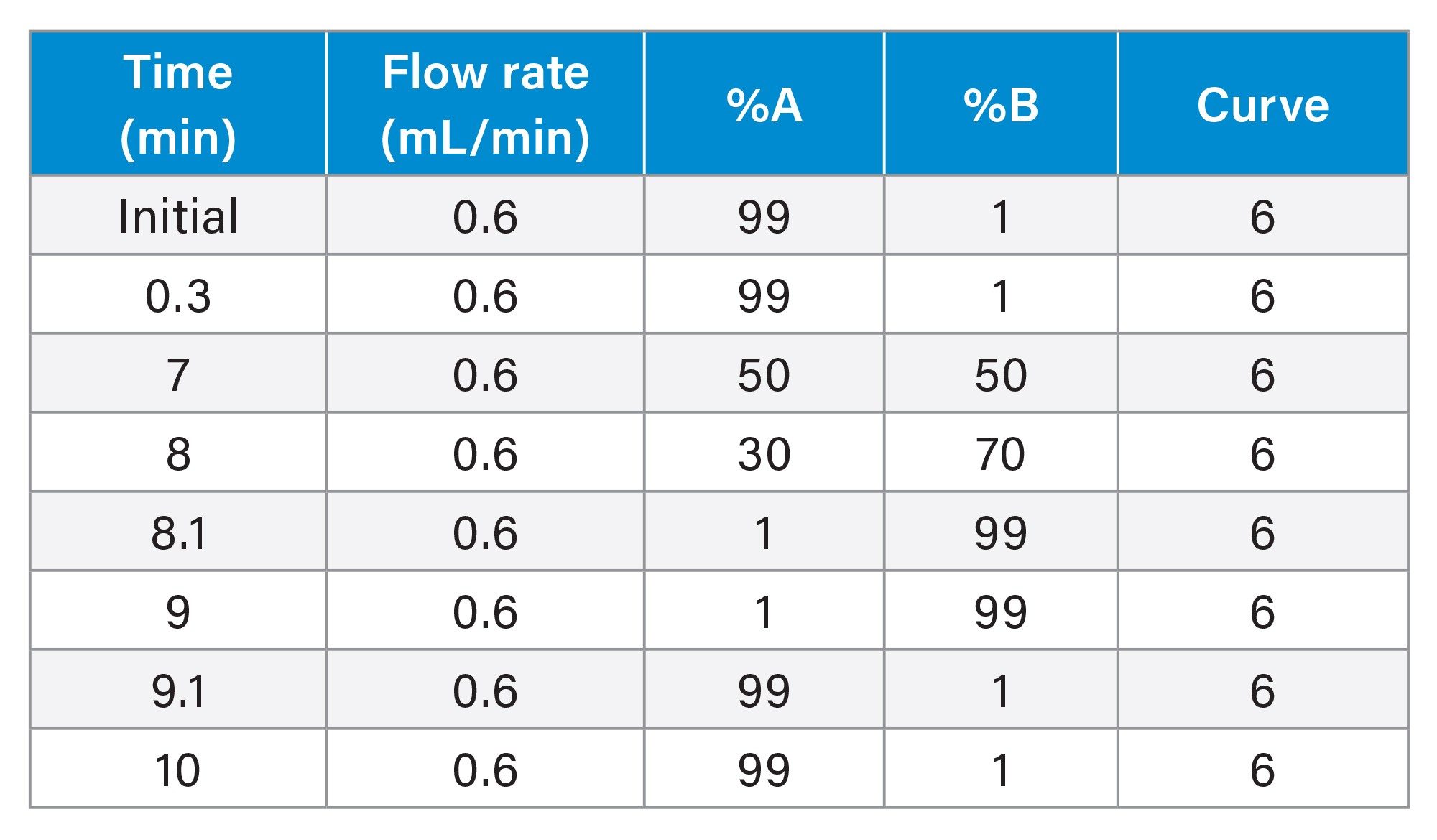
Gradient Table
MS Conditions
|
MS system: |
SELECT SERIES MRT |
|
Ionization mode: |
Positive and Negative ESI |
|
Acquisition range: |
50–2400 m/z |
|
Acquisition mode: |
MSE |
|
Instrument mode: |
MRT |
|
Capillary voltage: |
3.0 kV |
|
Collision energy ramp: |
10–40 eV |
|
Cone voltage: |
40 V |
Data Management
|
Chromatography software: |
MassLynx™ V4.2 |
|
MS software: |
MassLynx V4.2 |
|
Informatics: |
Progenesis QI V3.0 |
Results and Discussion
Data Acquisition and Processing
Each pooled urine sample was analyzed in triplicate with the pooled quality control sample analyzed at the beginning and end of the batch with injections interspersed throughout the sample list. To start the batch, a system suitability test injection of the Waters LCMS QC reference standard was acquired to assess the instrument performance including retention time and mass accuracy. This was followed by injections (n=10) of the QC sample to condition the system prior to analysis of the study samples. All the SELECT SERIES MRT data was acquired in both positive and negative ionisation in MRT mode by MSE with lockmass correction carried out using 200 pg/µL leucine enkephalin. The resulting raw datafiles were imported into Progenesis QI software for processing where the samples underwent run alignment, peaking picking and normalization. Picked features that had a %CV more than 30% in the QC samples were tagged and then hidden from further statistical analysis.
Statistical Analysis
During metabolomic studies, it is pivotal that the resulting LC-MS data is reproducible so that variance in the detected features are as a result of real differences in the samples rather than that resulting from analytical variation. The resulting peak list generated by Progenesis QI software was imported into Metaboanalyst for statistical analysis.3–4 The plots in Figure 1 show the results from principal component analysis (PCA) where good separation is seen between the different sample groups and the pooled QC sample in dark blue is tightly clustered as are the repeat injections from each of the different groups. This demonstrated good instrument stability (mass accuracy, retention time and signal intensity) and indicated a robust performance of the analytical system over the batch of injections. This robustness reduces analytical variation allowing biological variation to be fully explored.
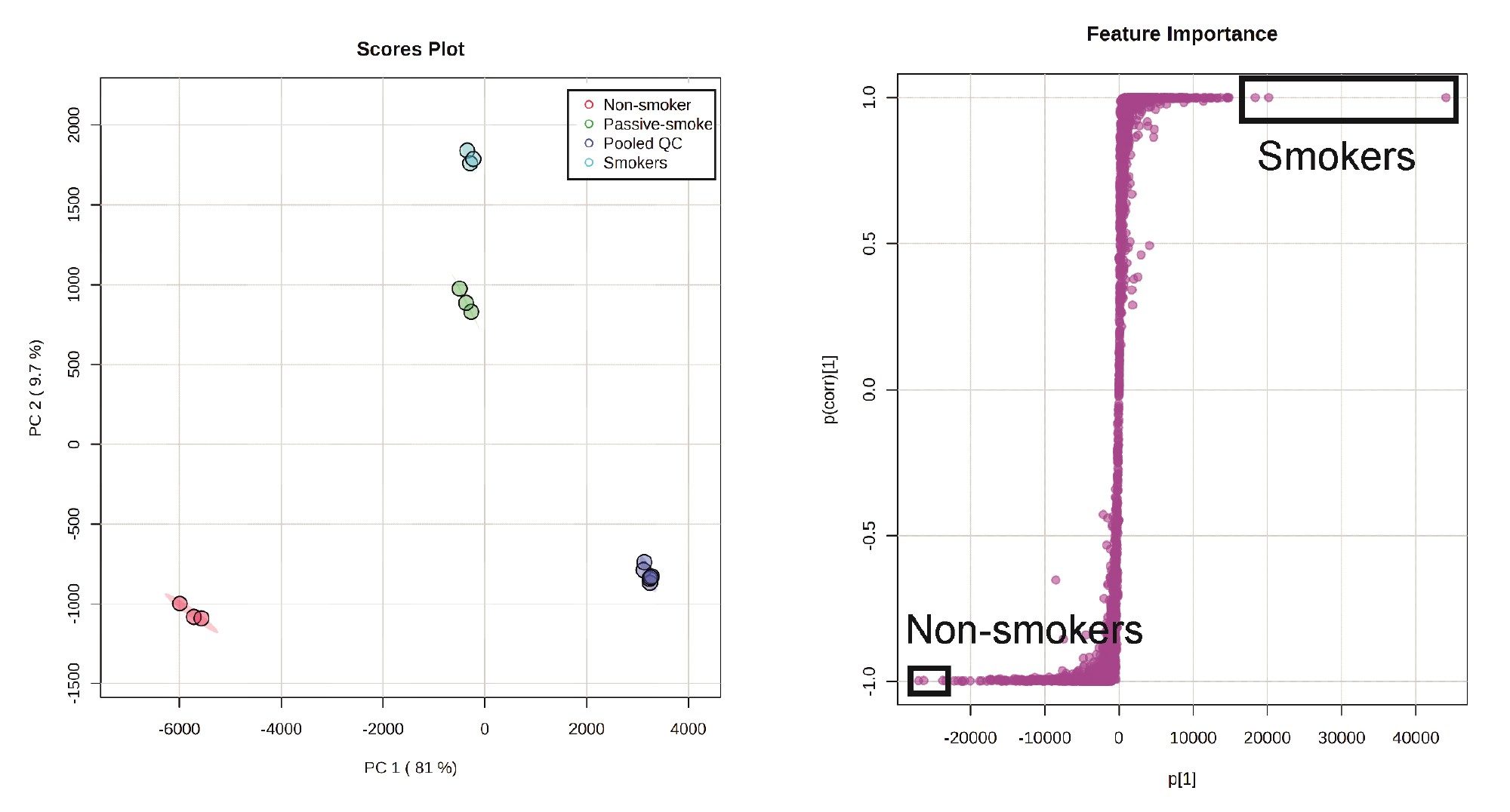 Figure 1. Principle component analysis of NIST urine samples (Left) and S-plot from OPLS-DA comparison between smokers and non-smokers with the top three significant features highlighted from each group.
Figure 1. Principle component analysis of NIST urine samples (Left) and S-plot from OPLS-DA comparison between smokers and non-smokers with the top three significant features highlighted from each group.
Following assessment of the group separations by PCA, binary group comparisons were performed using orthogonal partial least squared discriminate analysis (OPLS-DA). OPLS-DA is used for diagnosing the differences between two different groups of samples. It allows assessment of which feature loadings (accurate mass retention time pairs) contribute specifically to the variation between the two groups of interest. This is visualized as an S-plot (Figure 1) where the features that contribute to the biggest differences between the two groups are found. All three groups were analyzed by OPLS-DA (smokers vs non-smokers, smokers vs passive smokers and non-smokers vs passive smokers) where the top three significant features in each comparison as an example highlighted in the black boxes in Figure 1’s S-plot.
The box whisker plots shown in Figure 2 highlight the significance and normalized abundance of the selected features between smokers and non-smokers outlining these features as being major contributors to the variation between these study groups.
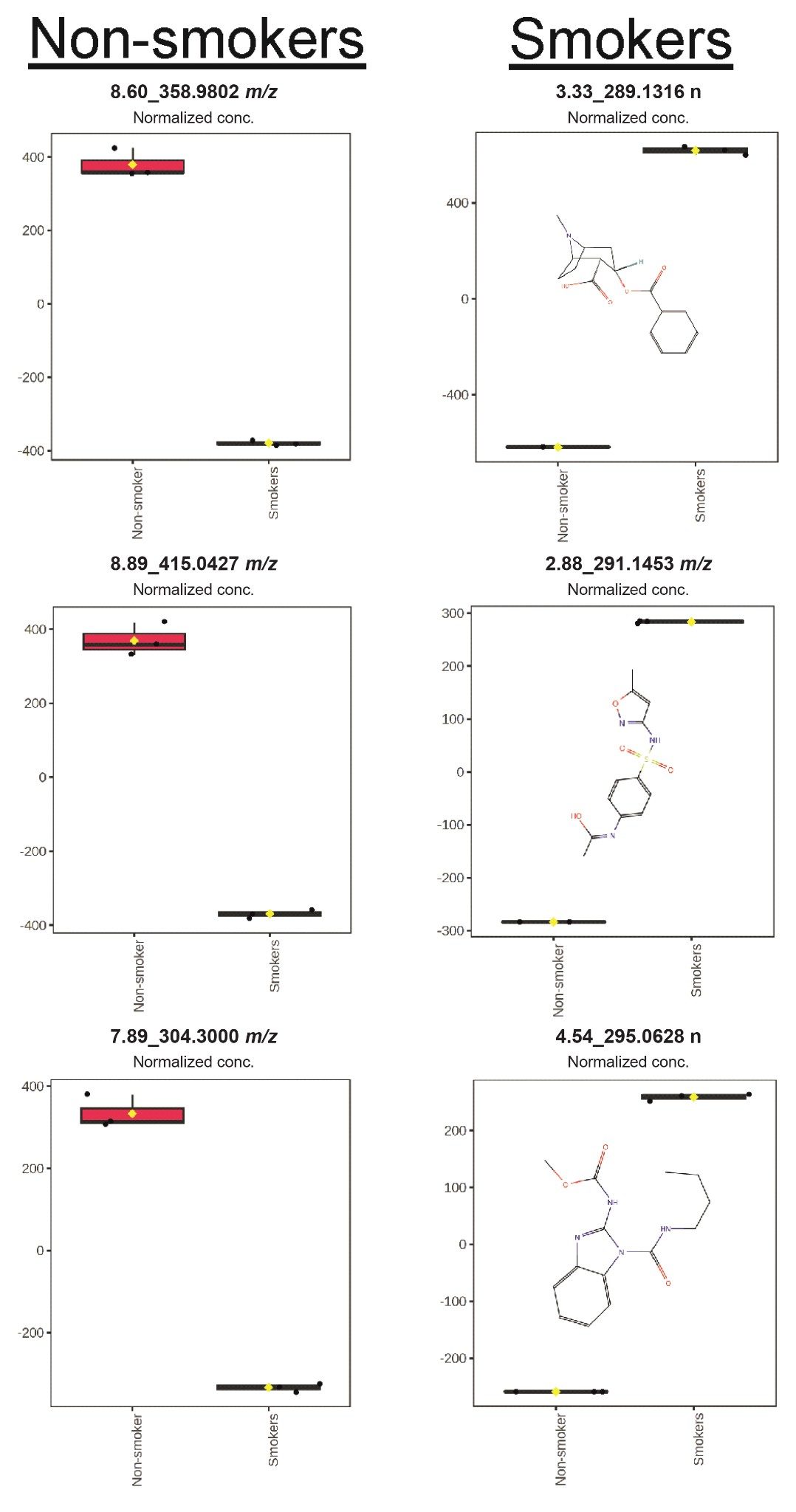 Figure 2. Box and whisker plots of the top three features determined from OPLS-DA for non-smokers and smokers.
Figure 2. Box and whisker plots of the top three features determined from OPLS-DA for non-smokers and smokers.
Confident Compound Annotation Using Ultrahigh Accurate Mass
Accurate sub ppm mass measurements provided by the SELECT SERIES MRT are important in providing confidence in compound annotations from available databases and libraries. The confidence in annotation can be further increased with the combination of accurate MS/MS fragment ion information. The top differentiated features determined by OPLS-DA were searched against databases including HMDB and an in-house FDA database for pharmaceuticals using precursor and fragment ion tolerances of 1 ppm with the resulting annotations outlined in Table 1. After the database searches, many of the resulting top hits were found to correspond to drugs or metabolites of drugs, including acetaminophen glucuronide, a widely used analgesic, ranitidine, a medication for treatment of peptic ulcers and reflux, acetyl sulfamethoxazole and trimethoprim, antibiotics administered as co-antibiotic (Cotrimoxazole) to treat lung infections linked to COPD and recreational drug cocaine and its biomarker metabolite benzoyl ecgonine.5–6 Four features could not be identified from the databases used. However, two were assigned tentative elemental compositions, and spectral examination of the remaining two features highlighted the potential presence of a dichloride isotopic pattern with the molecules potentially resulting from a metabolic conjugation of a halogen containing pharmaceutical drugs like diclofenac.
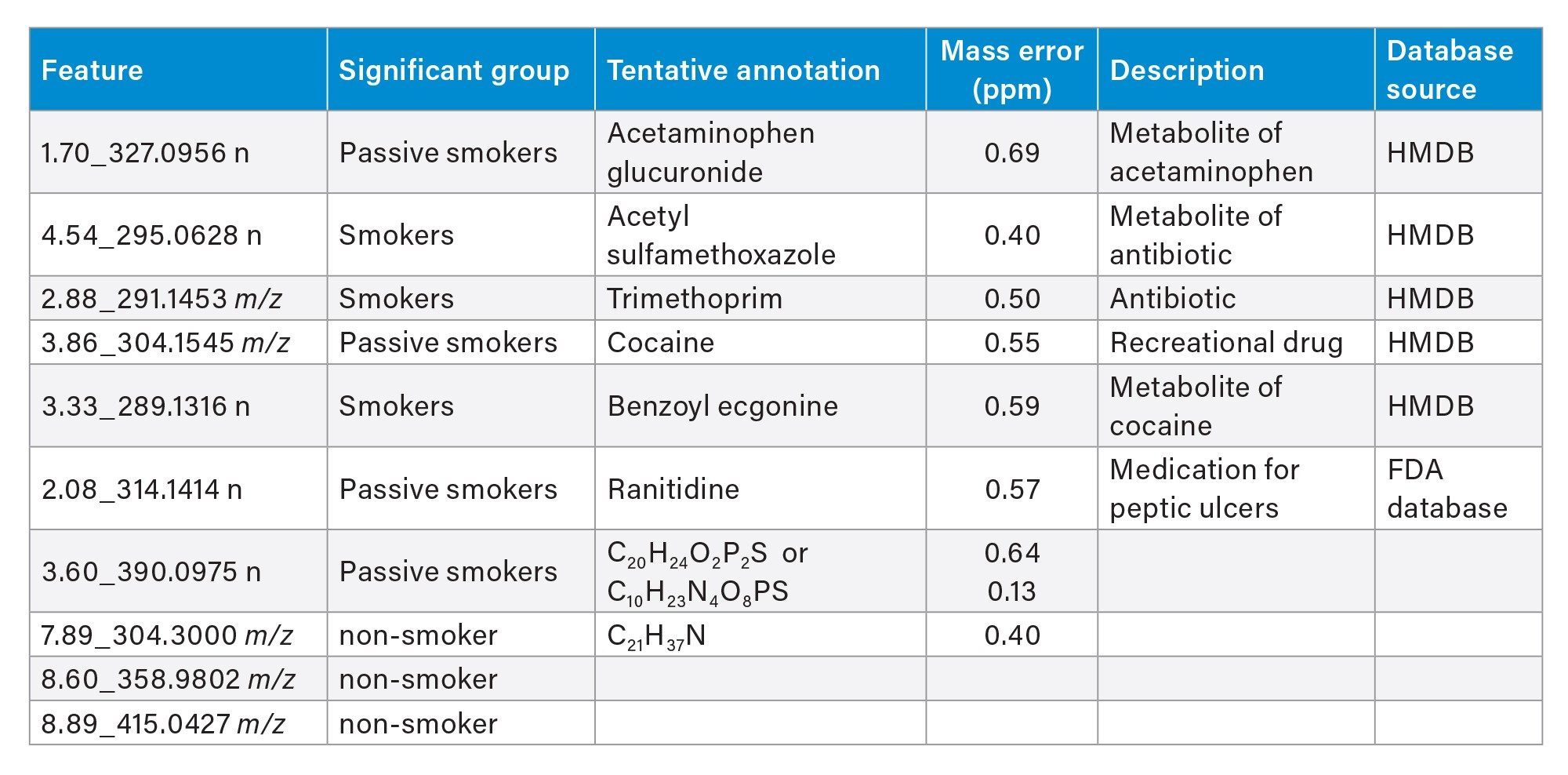 Table 1. List of Progenesis QI annotations of top three significant features from group OPLS-DA analysis.
Table 1. List of Progenesis QI annotations of top three significant features from group OPLS-DA analysis.
Figure 3 presents two examples of tentative annotations listed in Table 1 with their corresponding matching of In-silico fragmentation, mass accuracy and isotope similarity. The data provides high confidence that a correct assignment has been made for example for feature 3.33_289.1316n annotated as Benzoyl ecgonine shows a sub ppm mass error of 0.59 ppm and an isotope similarity of more than 99%. Additionally, several In-silico fragments were matched to the spectral fragment ions with the mass accuracy of these fragment ions each being less than 2 ppm.
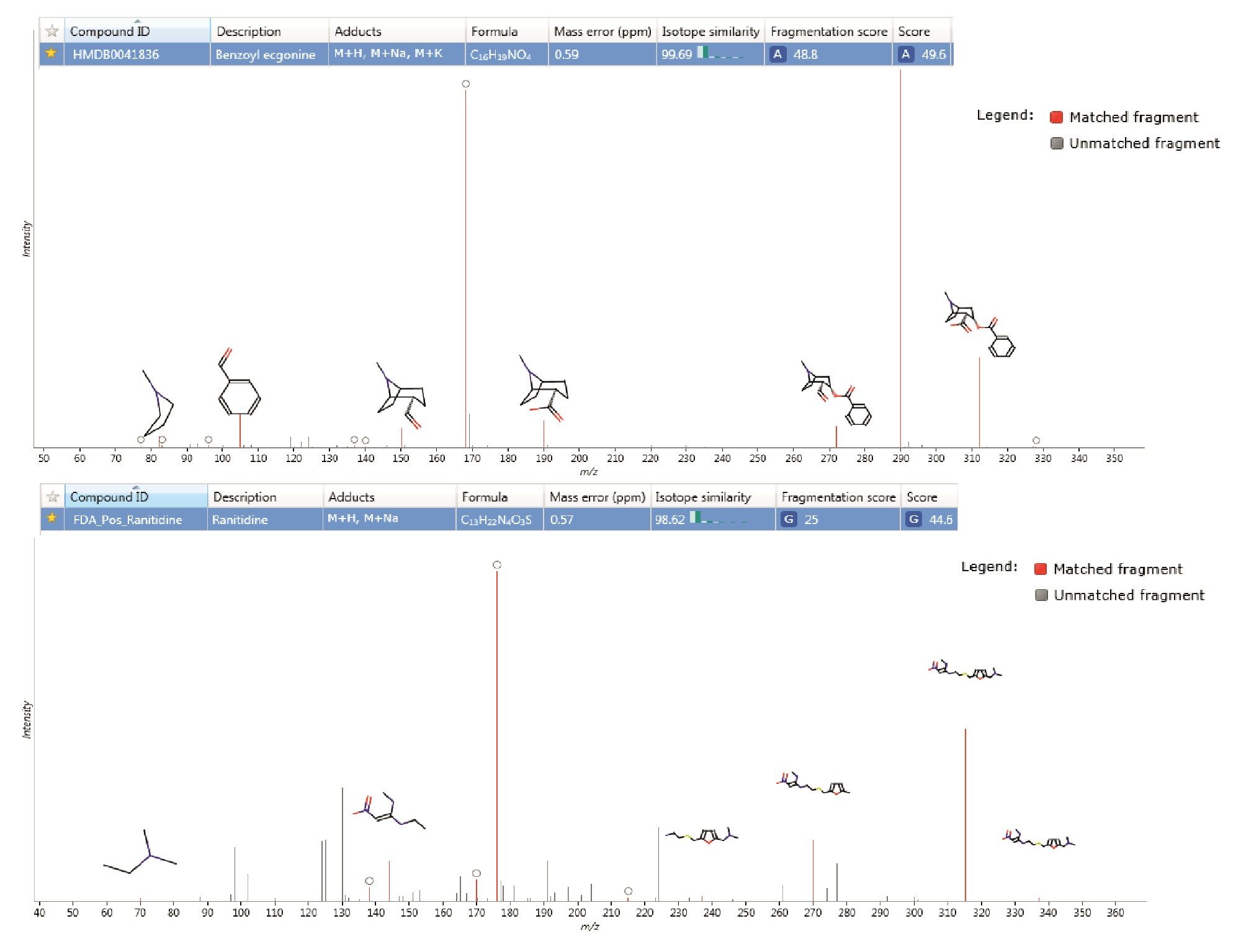 Figure 3. Tentative compound annotation from database searching in Progenesis QI showing matching of In-silico fragmentation.
Figure 3. Tentative compound annotation from database searching in Progenesis QI showing matching of In-silico fragmentation.
In addition to the improved annotation confidence provided by the accurate mass measurement and detailed fragmentation matching, examination of the isotopic pattern and finer isotopic distribution compared to theoretical elemental composition, can provide further confidence in accurate annotations. The spectra highlighted in Figure 4 shows an expanded region of the second isotopic peak cluster from the feature assigned as Benzoyl ecgonine (C16H20N1O4). The figure shows the theoretical isotopic profile in blue overlaid with the theoretical centroided masses in pink and finally centroided (lockmass corrected) raw data from the NIST urine analysis in green. As can be seen, the theoretical and experimental data align well with calculated mass errors for each ion ranging from 0.09 ppm to 1.86 ppm with almost identical relative abundances. The data in Table 2 further examines the relative abundance and reproducibility of the isotopes of this feature (3.33_289.1316n) compared again to the theoretical values. The relative percentage abundance of each main isotopic peak has been compared to the expected theoretical abundance and the reproducibility can be seen across three replicate injections where the percentage abundance is consistent. Additionally in the table, the reproducibility of the finer isotopic pattern outlined in Figure 4 was also compared across injections and to the theoretical and demonstrated good reproducibility for these low intensity ions.
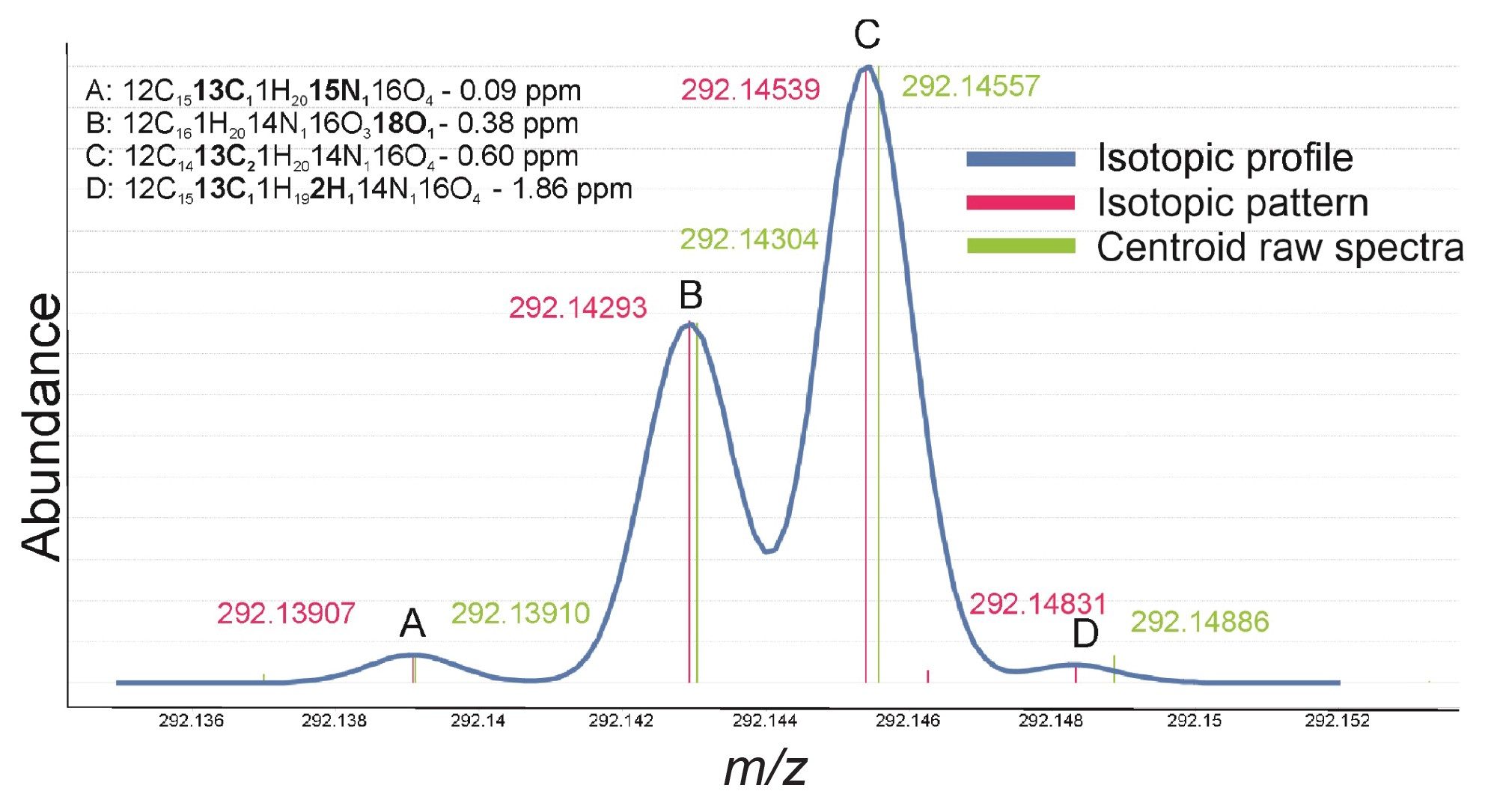 Figure 4. Example expanded spectra showing fine theoretical isotopic pattern (pink) of second isotope peak for feature 3.33_289.1316n annotated as benzoyl ecgonine, overlaid with centroided raw spectra (green) with inserted isotope mass accuracy to theoretical.
Figure 4. Example expanded spectra showing fine theoretical isotopic pattern (pink) of second isotope peak for feature 3.33_289.1316n annotated as benzoyl ecgonine, overlaid with centroided raw spectra (green) with inserted isotope mass accuracy to theoretical.
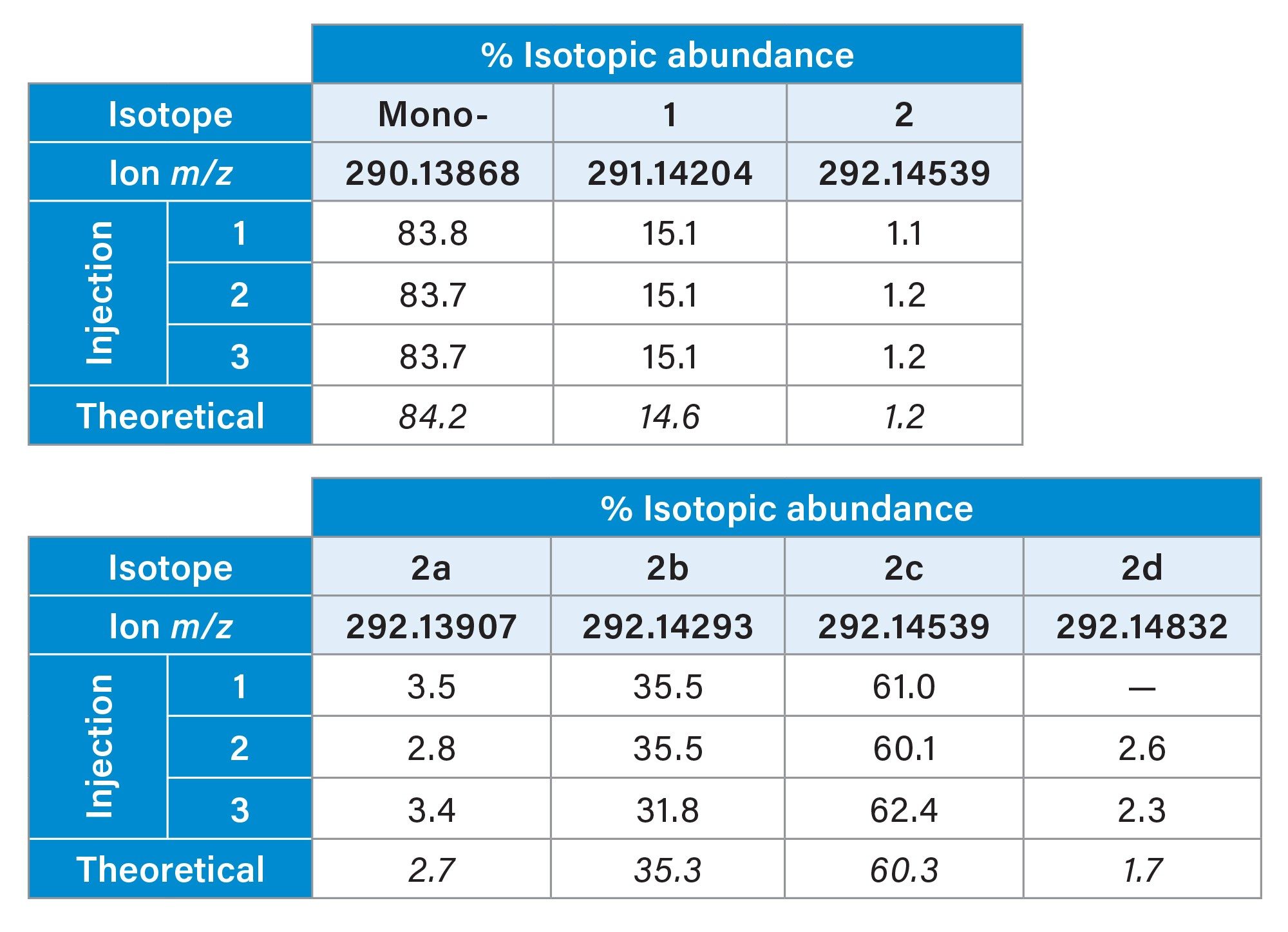 Table 2. Reproducibility of isotope abundance for main isotopic peaks of feature 3.33_289.1316n from the smoker sample (a) and fine isotopic ions compared to theoretical abundance (b).
Table 2. Reproducibility of isotope abundance for main isotopic peaks of feature 3.33_289.1316n from the smoker sample (a) and fine isotopic ions compared to theoretical abundance (b).
Once features of interest have been determined and annotated, normalized abundance profiles can be viewed in Progenesis QI to determine changes in levels of metabolites across sample groups. The profile plots shown in Figure 5 highlight the changes in features annotated as nicotine and its metabolites (including cotinine, Nicotine glucuronide, Cotinine glucuronide, hydroxycotinine, Nicotine N-oxide and Trans-hydroxycotinine glucuronide) across the three different sample groups. These abundance profiles can assist in increasing the confidence of the annotations as their abundance changes as expected across the groups with the smokers having the highest level of the nicotine metabolites, decreasing with the passive smoker group to a zero abundance with the non-smoker grouping. A miss identification of a nicotine metabolite would not follow this trend and could be ruled out.
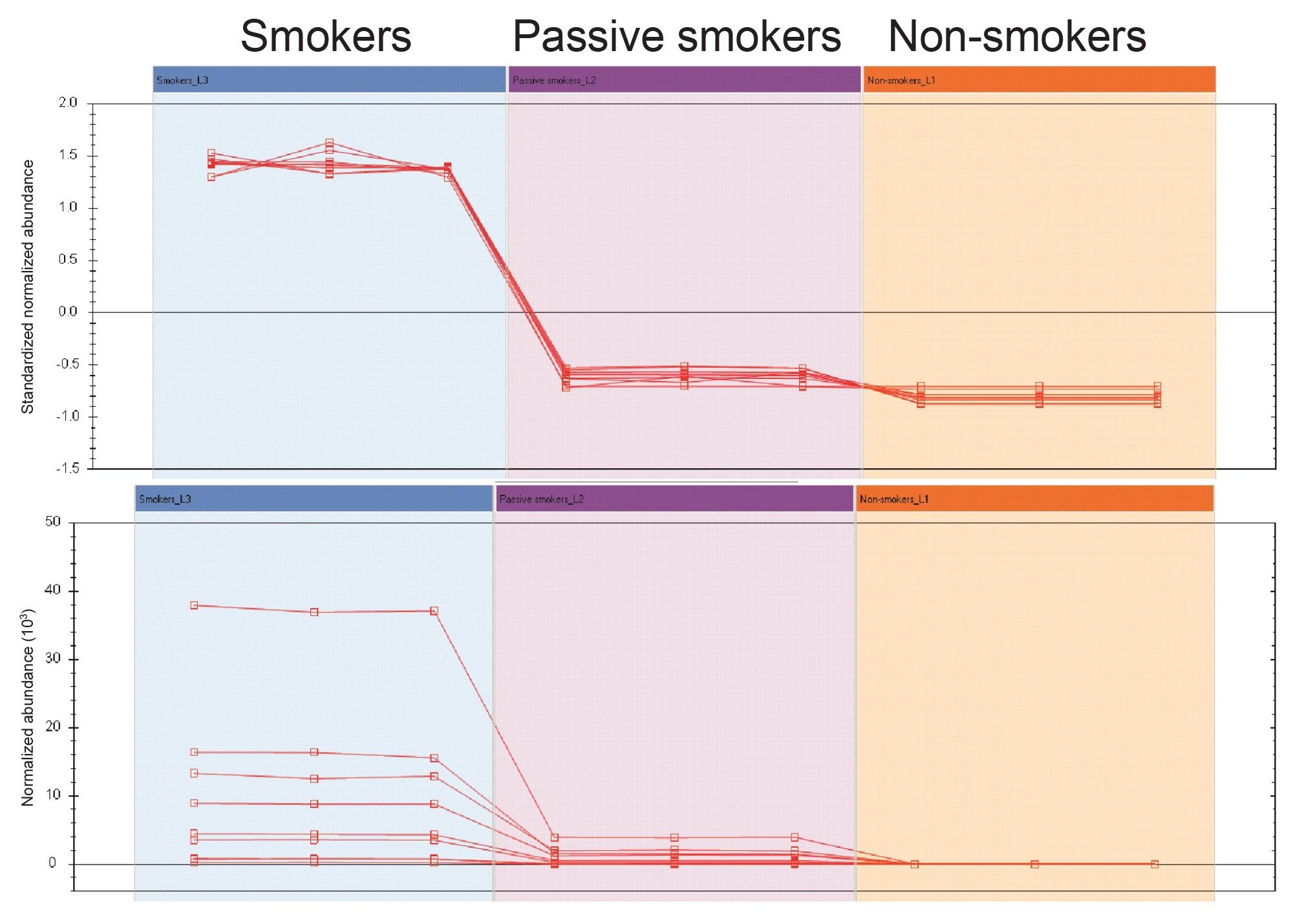 Figure 5. Abundance profile of nicotine metabolites across group sample injections.
Figure 5. Abundance profile of nicotine metabolites across group sample injections.
To fully understand the biological impact of smoking (or impact of drugs and pharmaceuticals), it is vital to accurately identify all of the exogenous features and their metabolites and remove them, so they do not influence further statistical analysis. The PCA plots highlighted in Figure 6 show the statistical impact on the removal of the exogenous features. Before their removal the smokers and passive smoker group separated along principal component two however with the removal of the nicotine and cocaine metabolites they are no longer separated. The statistics now indicate that the underlining biological mechanism impacting smokers also affects those individuals exposed to passive smoking.
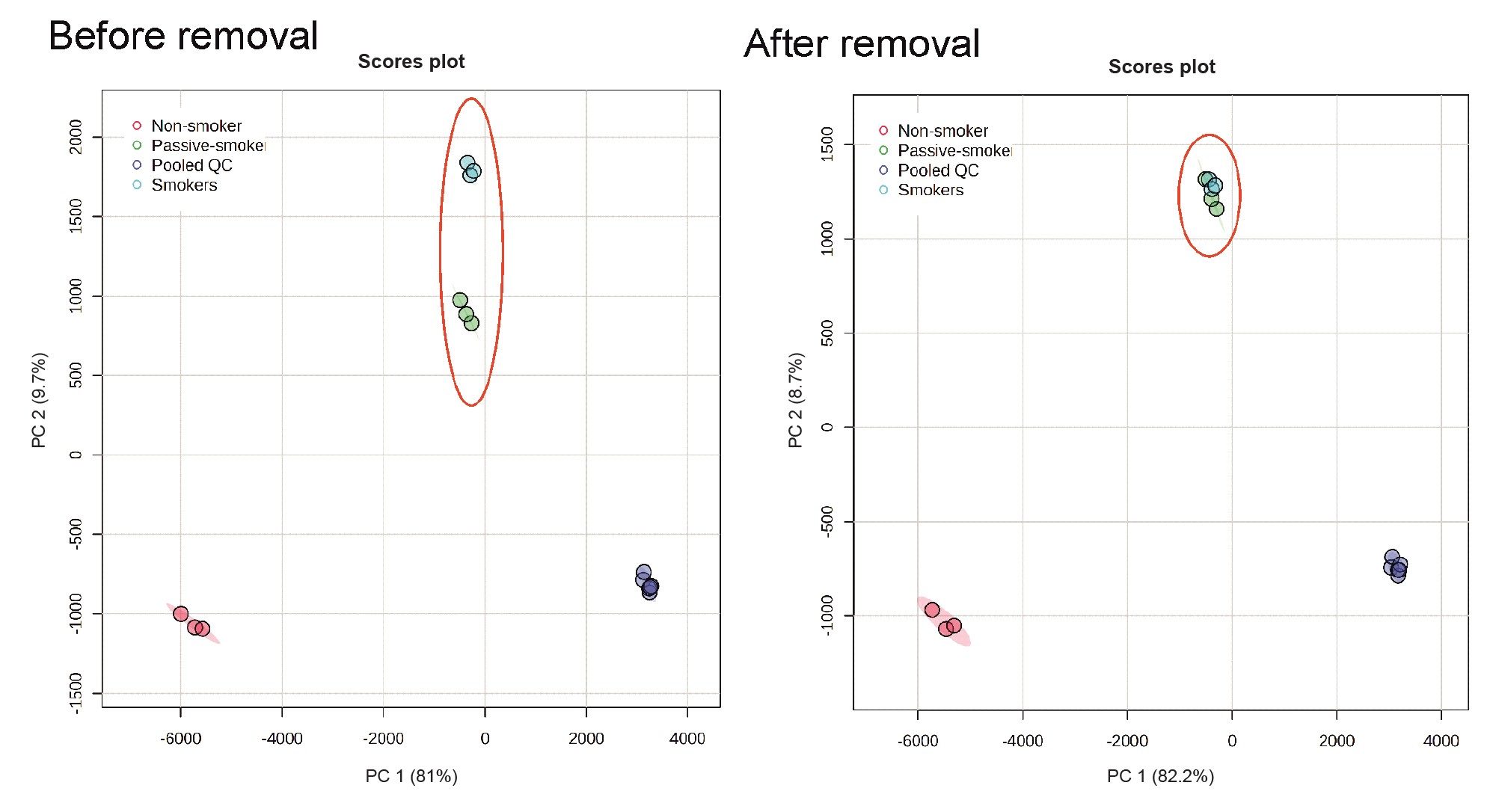 Figure 6. PCA plots of NIST urine sample and QCs for all features and after removal of features linked to pharmaceuticals and nicotine metabolites.
Figure 6. PCA plots of NIST urine sample and QCs for all features and after removal of features linked to pharmaceuticals and nicotine metabolites.
Conclusion
Discovery metabolomic studies routinely involve analysis of large numbers of samples and during these analysis low mass resolution and variability in mass accuracy can lower the confidence in identification of potential biomarkers, impacting downstream interpretation of the results. In this application note, we have highlighted how data generated on the SELECT SERIES MRT integrates with UPLC and the Waters metabolomic workflow and outlined how the unique capabilities of the instrument can enhance the quality of the data. The high mass accuracy and high mass resolution combined with quality MS/MS data greatly increased the accuracy and confidence in the annotation of significant features of interest between the urine samples. Further more, we highlight the importance of determining tentative identifications of analytes to improve the interpretation of relevant biological mechanisms.
References
- Smith, C. A, Want, E. J, O'Maille, G, Abagyan, R, Siuzdak, G. XCMS: Processing Mass Spectrometry Data for Metabolite Profiling Using Nonlinear Peak Alignment, Matching, and Identification. Analytical Chemistry 2006, 78 (3), 779–787.
- Cooper-Shepherd, D. A, Wildgoose, J, Kozlov, B, Johnson, W. J, Tyldesley-Worster, R, Palmer, M. E, Hoyes, J. B, McCullagh, M, Jones, E, Tonge, R, Marsden-Edwards, E, Nixon, P, Verenchikov, A, Langridge, J. I. Novel Hybrid Quadrupole-Multireflecting Time-of-Flight Mass Spectrometry System. J Am Soc Mass Spectrom 2023, 34 (2), 264–272.
- Pang, Z, Zhou, G, Ewald, J, Chang, L, Hacariz, O, Basu, N.; Xia, J. Using MetaboAnalyst 5.0 for LC-HRMS Spectra Processing, Multi-Omics Integration and Covariate Adjustment of Global Metabolomics Data. Nat Protoc 2022, 17 (8), 1735–1761.
- Xia, J, Psychogios, N, Young, N, Wishart, D. S. MetaboAnalyst: A Web Server for Metabolomic Data Analysis and Interpretation. Nucleic Acids Res 2009, 37 (Web Server issue), W652–60.
- Mwenya, D. M, Charalambous, B. M, Phillips, P. P, Mwansa, J. C, Batt, S. L, Nunn, A. J, Walker, S, Gibb, D. M, Gillespie, S. H. Impact of Cotrimoxazole on Carriage and Antibiotic Resistance of Streptococcus Pneumoniae and Haemophilus Influenzae in HIV-infected Children in Zambia. Antimicrob Agents Chemother 2010, 54 (9), 3756–62.
- Sohy, C, Pilette, C, Niederman, M. S, Sibille, Y. Acute Exacerbation of Chronic Obstructive Pulmonary Disease and Antibiotics: what studies are still needed? Eur Respir J 2002, 19 (5), 966–75.
Featured Products
720008296, April 2024