Automated Method Development Using Analytical Quality-by-Design for Stability Indicating Methods
Abstract
New antiviral therapies with increased potency, greater genetic barriers to resistance, and improved long-term safety and tolerability are rapidly being developed to provide treatment for conditions such as COVID-19. Chromatographic stability indicating methods (SIM)s are employed to monitor the assay, degradation products and impurities of these therapeutic compounds. The utility of an automated and reliable AQbD approach to the development of chromatographic SIMs can provide robustness and efficiency advantages when developing methods using forced degradation sample preparations.1
Benefits
- Stability indicating methods can be developed in an automated fashion using the Fusion QbD Method Development Software.
- The forced degradation feature in the Fusion QbD Method Development Software provides the ability to include multiple forced degradation samples in an automated method development design.
Introduction
Knowledge regarding the stability of active pharmaceutical ingredient (API)s is critical for suitable formulation, packaging selection, and shelf-life determination.2,3 Development of robust and reproducible analytical methods can be challenging, time consuming, and complex. Strategies routinely employed include: one-factor-at-a-time (OFAT), systematic protocol, and analytical quality-by-design (AQbD).
AQbD has recently gained attention from regulatory agencies, as the information gained with this approach supports knowledge-based decision making. By performing a risk assessment, data collected using AQbD principals generates a design space known as the Method Operable Design Region (MODR). From within this space, the acceptable performance region (APR) further defines the working boundaries for which method variables (i.e. temperature, gradient time, flow rate, etc.) meet specified system suitability requirements.1 Method operation within this boundary maximizes chromatographic robustness, while minimizing the likelyhood of aberrant, out-of-specification (OOS) or out-of-trend (OOT) results due to routine, day-to-day chromatographic variability.
In this study, Fusion QbD Method Development Software was employed in conjunction with Empower 3 to develop a stability indicating method (SIM). This method was used to monitor Tenofovir alafenamide fumarate (TAF), an antiviral prodrug and potent inhibitor of viral reverse transcription for treatment of chronic Hepatitis B and HIV.4 The “Forced Degradation” feature built into the Fusion QbD Software was used to include multiple forced degradation sample preparations in a single design space.
Experimental
Materials and Forced Degradation Preparation
Tenofovir alafenamide fumarate (TAF) certified reference material (CAS 379270-37-8) (Figure 1) (Selleckchem.com, Houston, TX, USA) was prepared at approximately 0.5 mg/mL in diluent (50/50 water/methanol). Four unique TAF forced degradation sample preparations were made to contain a final concentration of 5 mM HCl (acid), 0.025 mM ammonium formate pH 8.5 (base), 5% H2O2 (oxidation), and sample diluent (control). The control and oxidation samples were exposed to 60 °C for 3 hours, while samples containing acid and base were stored at room temperature for 3 hours prior to analysis.
 Figure 1. Structure of Tenofovir alafenamide fumarate.(5)
Figure 1. Structure of Tenofovir alafenamide fumarate.(5)
LC Conditions
|
LC system: |
ACQUITY Arc System with Quaternary Solvent Manager (rQSM), Sample Manager (rFTN), Column Manager (rCM) |
|
Detection: |
ACQUITY Photodiode Array Detector (PDA), UV 260 nm |
|
Column(s): |
XBridge Premier BEH Shield RP18, 2.5 µm, 4.6 x 150 mm, p/n: 186009923 XBridge Premier BEH C18, 2.5 µm, 4.6 x 150 mm, p/n: 186009849 XSELECT Premier CSH Phenyl-Hexyl, 2.5 µm, 4.5 x 150 mm, p/n: 186009888 XSELECT Premier CSH C18 XP, 2.5 µm, 4.5 x 150 mm, p/n: 186009874 |
|
Sample temp.: |
20 °C |
|
Injection volume: |
5.0 µL |
|
Flow rate: |
1.3 mL/min |
|
Aqueous mobile phase: |
10 mM Ammonium formate pH 4.0 10 mM Ammonium formate pH 6.4 10 mM Ammonium formate pH 8.4 |
|
Organic mobile phase: |
Acetonitrile |
Data Management
|
Data management: |
Empower 3 Chromatography Software and Fusion QbD Software from S-Matrix Corporation |
Results and Discussion
Method development using AQbD principals included a series of systematic phases. In this study, an initial design (Rapid Screen) was developed and executed for the four forced degradation sample preparations. After integration and statistical processing of chromatograms generated in the MODR, a second design to explore more focused variables (Method Optimization) was developed and executed. From this, the APR was defined and method performance within this region was verified to finalize conditions for the SIM. The systematic method development phases are further described in detail below.
Phase 1: Rapid Screening
Mobile phase pH, column, and gradient time variables were included in the Fusion QbD rapid screening method development design. The “Replication Settings/Forced Degradation Study” feature (Figure 2) was employed to include all four forced degradation sample preparations (acid, base, oxidation, control) in the screening process. The design was then imported into Empower 3 for implementation.
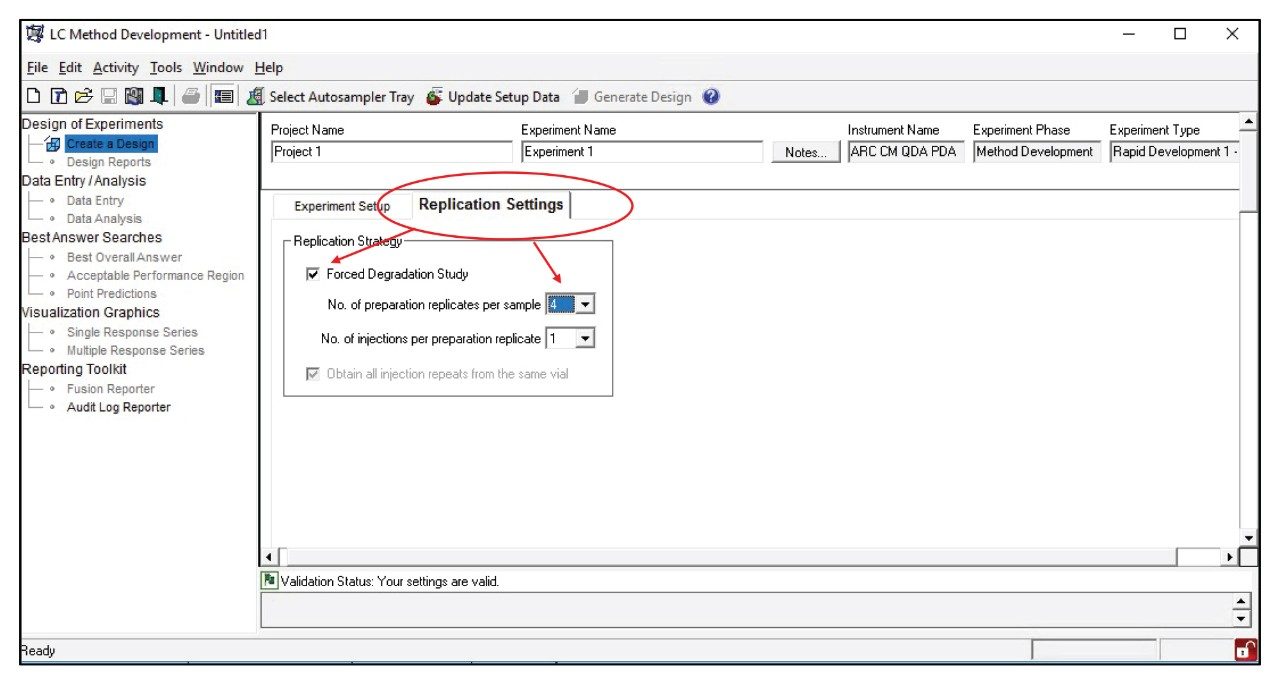 Figure 2. LC method development settings in the Fusion QbD Software. The four forced degradation sample preparations were included in the study design via the Replication Settings tab.
Figure 2. LC method development settings in the Fusion QbD Software. The four forced degradation sample preparations were included in the study design via the Replication Settings tab.
Phase 2: Importing Data into Fusion QbD and Developing a Knowledge Space
After run completion, all chromatograms were integrated using a generic processing method. The four degradation sample preparation chromatograms were visually overlaid for the first set of variables executed in the design. Although many degradation products were common between the four preparations, the oxidized sample preparation exhibited additional, unique degradation products. The oxidized sample preparation was selected for statistical analysis and further method development, acting as a “worst-case” scenario for the number of potential peaks.
Integrated chromatograms for the oxidized sample preparation injections were imported into Fusion QbD for statistical processing. A “peak count” performance goal was selected to generate trend response information. From this, mathematical models were automatically built by the software based upon the peak profile present in the imported chromatograms. Fusion QbD predicted the method variable combinations (pH, column, gradient time) that would generate the “Best Overall Answer (BOA)” or maximized peak count. These variables were carried forward into method optimization after completion of the rapid screening study.
Phase 3: Method Optimization – Temperature and Gradient Time
The method optimization design was built to explore column temperatures between 40–50 °C, with further refinement of the gradient time. After design execution, chromatogram integration, and data import into Fusion QbD, the acceptable performance region (APR) was defined by maximizing chromatographic system suitability performance goals including; peak count, USP resolution, and k’, while at the same time, minimizing USP tailing (Figure 3).
The APR predicted that a gradient time between 7–9 minutes and a column temperature between 41.5–43.5 °C would provide a robust chromatographic separation. System suitability criteria for peak count, USP resolution, k’, and USP tailing were all met within the APR region.
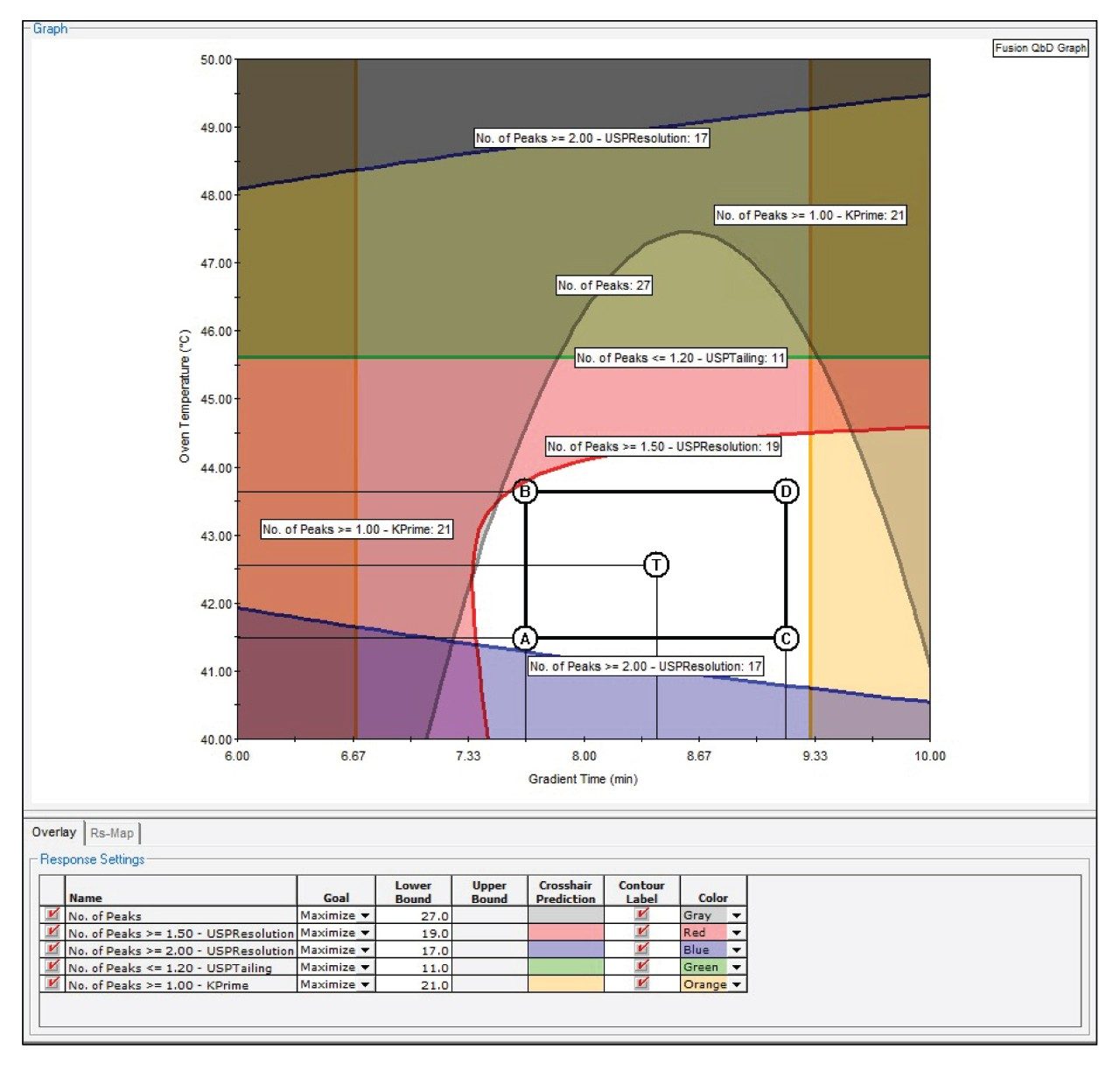 Figure 3. Fusion QbD graph of the design space and the APR region obtained from the method optimization experiment. The table under the graph shows the user-specified performance goals that were achieved in this design.
Figure 3. Fusion QbD graph of the design space and the APR region obtained from the method optimization experiment. The table under the graph shows the user-specified performance goals that were achieved in this design.
Phase 4: Verification
Method verification was performed by analyzing all four forced degradation sample preparations under the variables defined at the central point within the APR. As per the rapid screening result, the XBridge Premier BEH C18 Column was employed with 10 mM ammonium formate pH 4.0, and as per the method optimization study, the column temperature was operated at 43 °C with a 2 minutes hold at 2% B. The gradient was then increased to 95% over 8.5 minutes. The flow rate, injection volume, and UV detection wavelength were held constant throughout the study at 1.3 mL/min, 5 µL, and 260 nm, respectively (Figure 4).
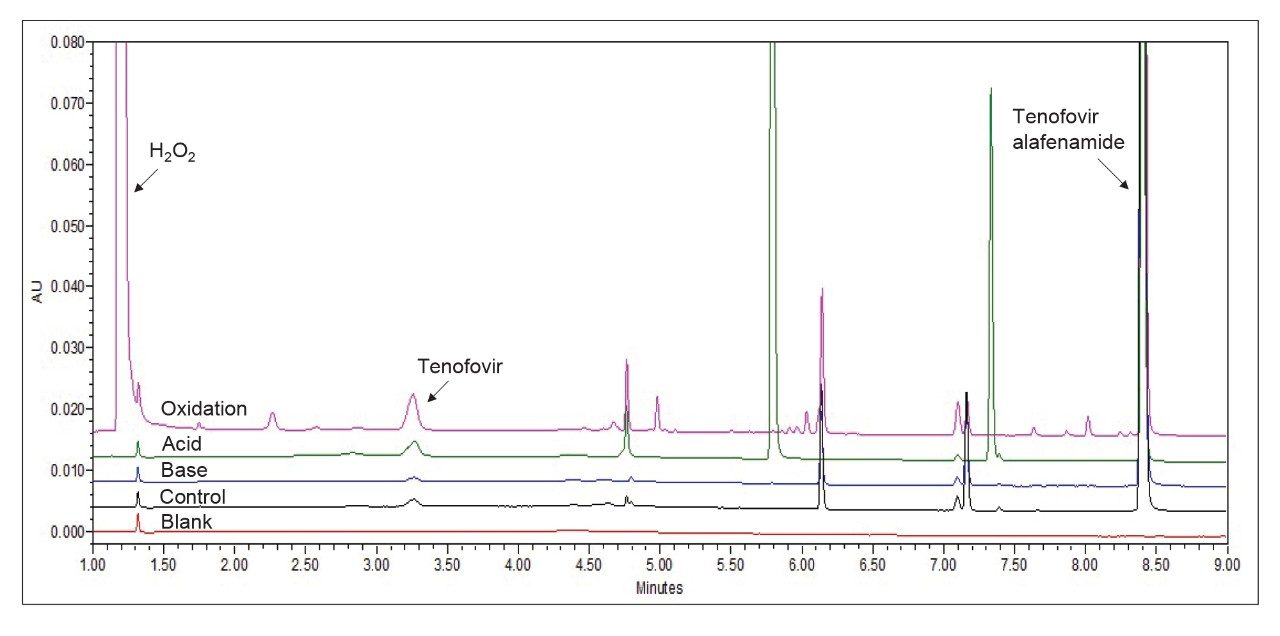 Figure 4. Final SIM chromatographic profile for Tenofovir alafenamide fumarate forced degradation sample preparations.
Figure 4. Final SIM chromatographic profile for Tenofovir alafenamide fumarate forced degradation sample preparations.
Conclusion
In this study, we demonstrated the development of a stability indicating method used to monitor Tenofovir alafenamide fumarate drug substance after exposure to forced degradation conditions. An automated, statistical approach was employed by utilizing Fusion QbD Method Development Software. This approach provided the ability to explore the chromatographic performance of multiple sample preparations within an automated method development design.
References
- Fadi L. Alkhateeb, Paul D. Rainville. “Analytical Quality by Design Based Method Development for the Analysis of Formoterol, Budesonide, and Related Compounds Using UHPLC-MS”. Waters Application Note, 2019, 720006696.
- “Guidance for Industry #5, Drug Stability Guidelines, FDA Code of Federal Regulations Title 21, Volume 4 (21CFR211), accessed 11/16/21.
- ICH guidelines, Q1A (R2): Stability Testing of New Drug Substances and Products (revision 2), International Conference on Harmonization, 2003.
- Vijaya Madhyanapu Golla, Moolchand Kurmi, Karimullah Shaik, Saranjit Singh. “Characterization of Degradation Products of Tenofovir Alafenamide Fumarate and Comparison of Its Degradation and Stability Behaviour With Tenofovir Disoproxil Fumarate”. Journal of Pharmaceutical and Biomedical Analysis 131 (2016) 146–155.
- 2D Structure Database, www.ChemSpider.com, accessed 10/20/21.
720007480, December 2021