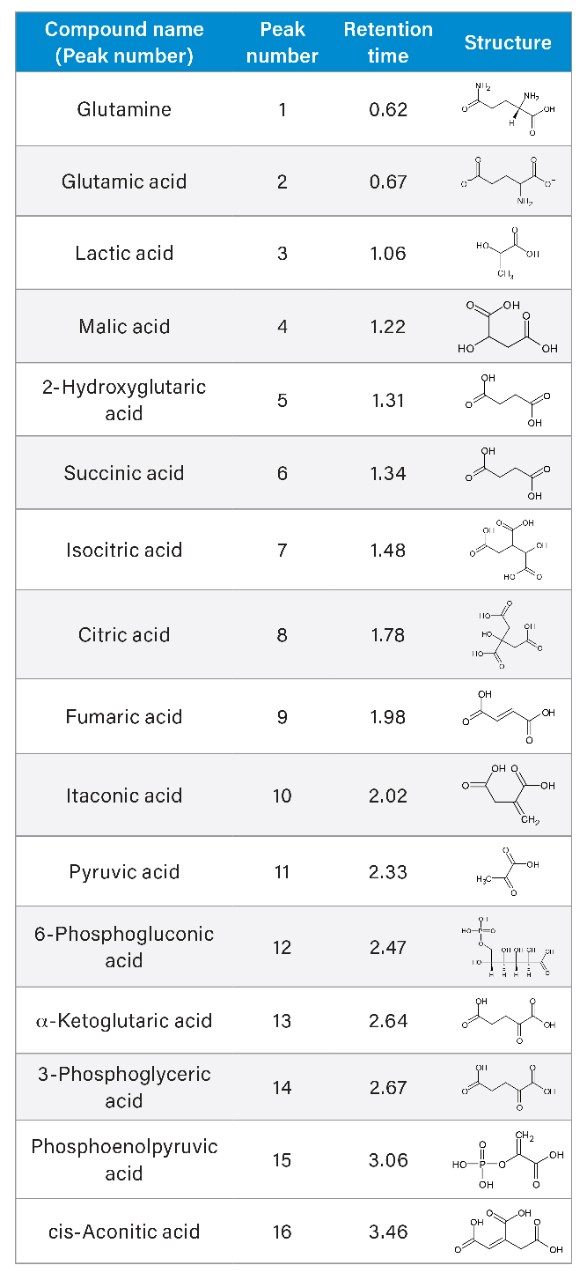
The tricarboxylic acid (TCA) cycle, also known as the Krebs cycle or Citric acid cycle, is the ultimate fate of metabolism where Acetyl-CoA or other molecules are formed by the breakdown of carbohydrates, protein, and fats.1 These molecules are then enzymatically oxidized to produce molecules such as adenosine triphosphate (ATP) to fuel cellular growth and function, as well as to reduce important cofactors that enables other metabolic processes.1 In addition, the TCA cycle produces precursors for amino acids, proteins, fatty acids, cholesterols, and nucleotide synthesis for cell growth and division.2
Due to the essential metabolic processes and pathways where the TCA cycle is involved, there is a need to study the changes in the up and down regulation of analytes and how they can enable new understandings for disease states and cellular processes.3 The components of the TCA cycle are small and polar carboxylic acids making them difficult to retain under traditional reversed-phase LC conditions. Some of the current LC methods that are utilized to analyze these compounds include: HILIC,4 ion-pairing,5 anion exchange,6 and in addition derivatization followed by either gas7 or liquid chromatography.8 While each methodology has its own unique challenges, further complications can be caused by the presence of metal surfaces. Compounds with electron-rich moieties, such as carboxylates and phosphates, are known to chelate metals, notably iron.9
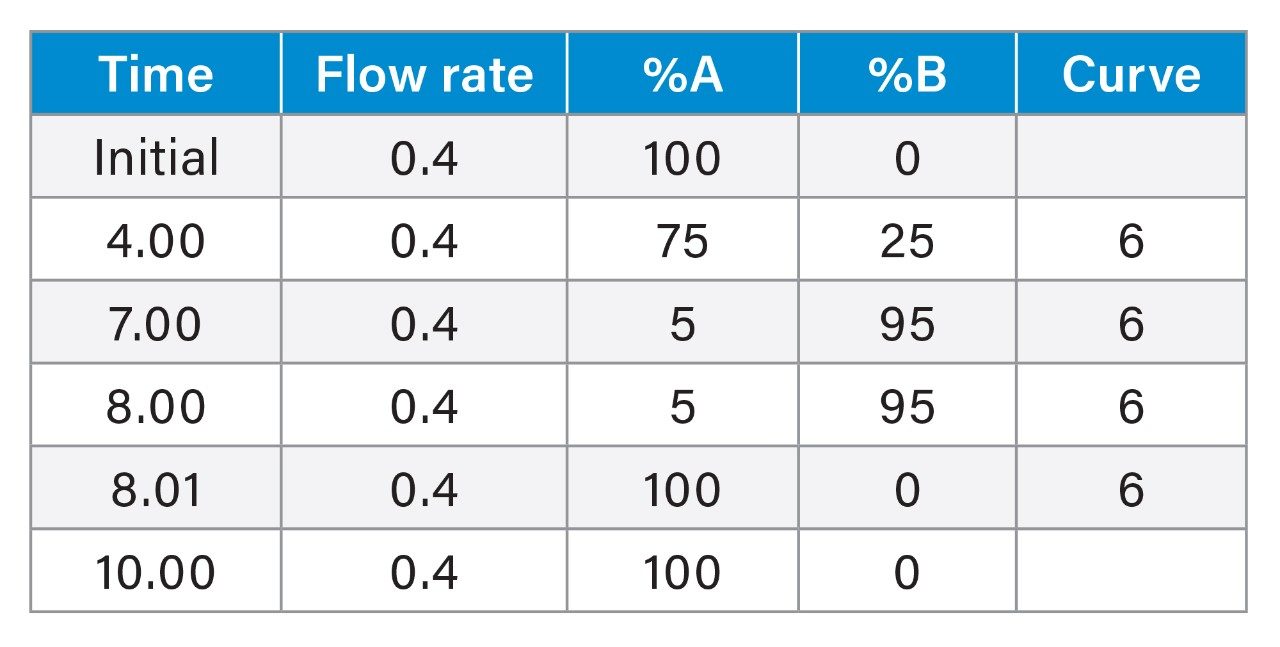
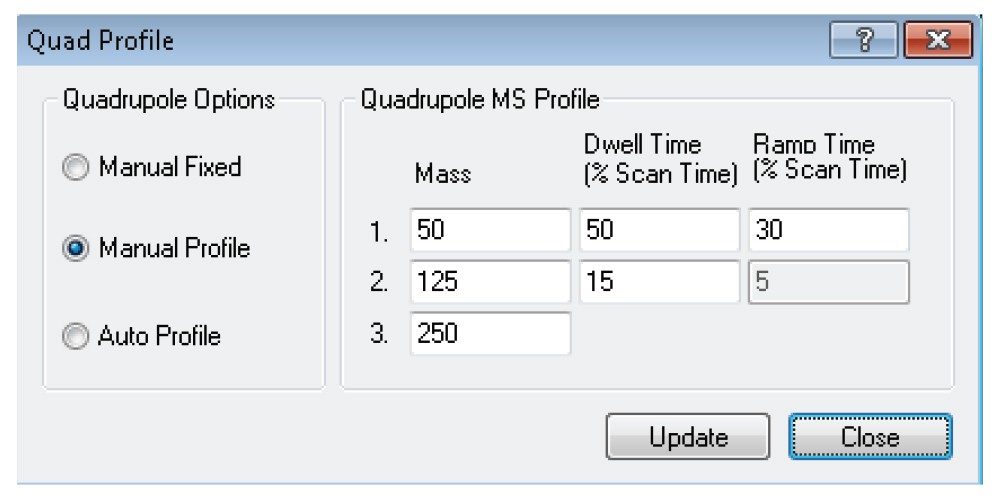
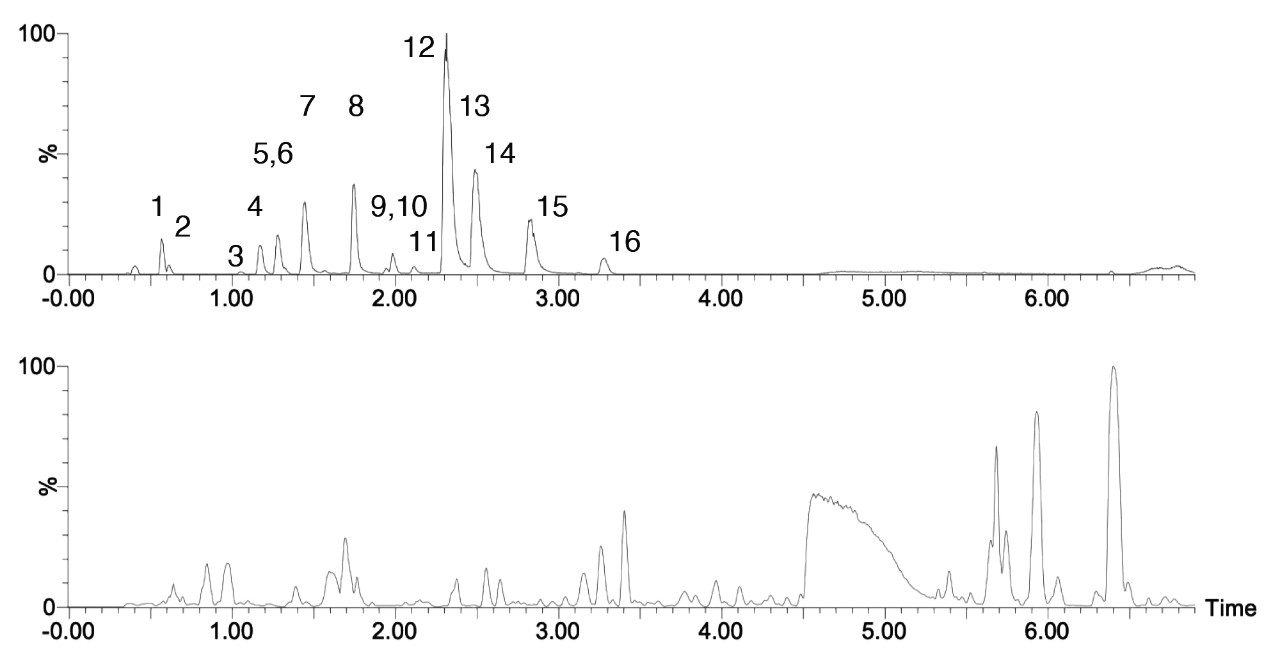
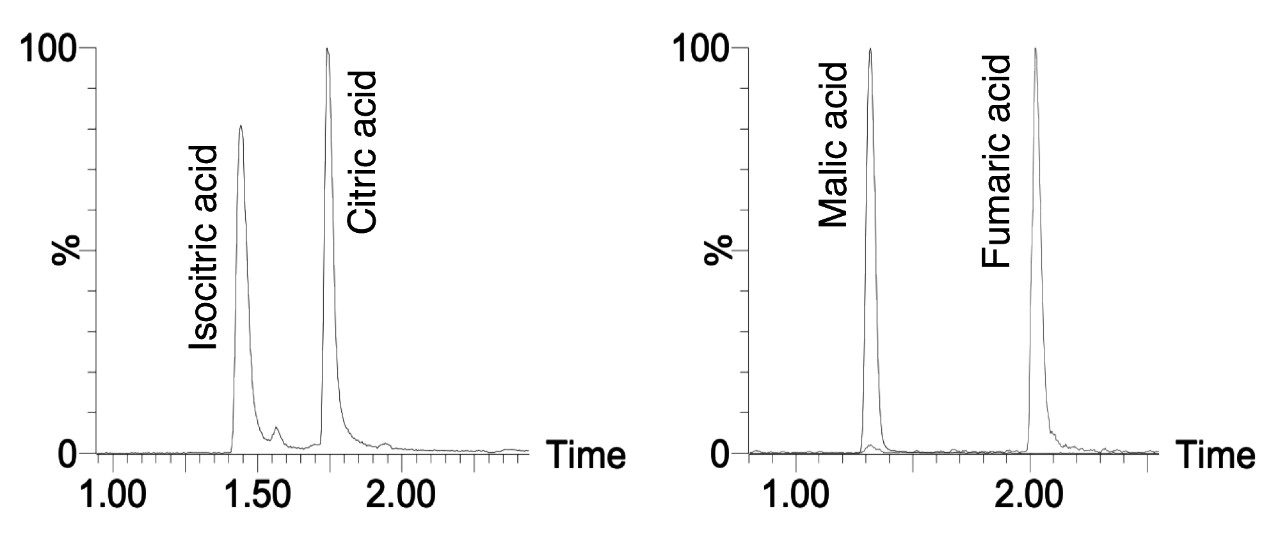
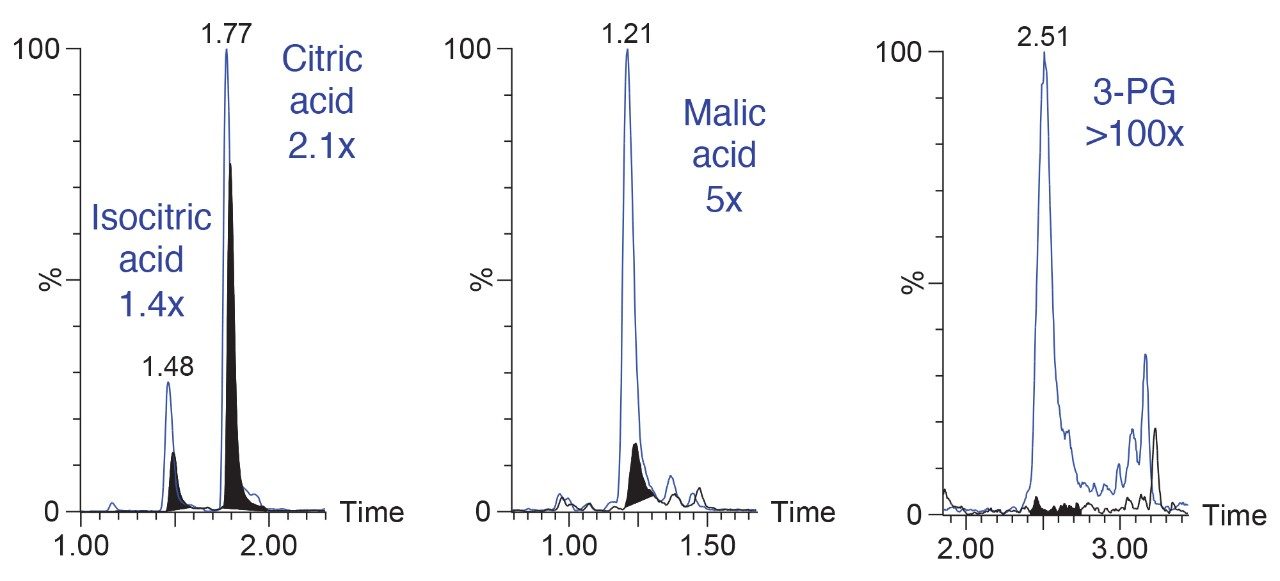
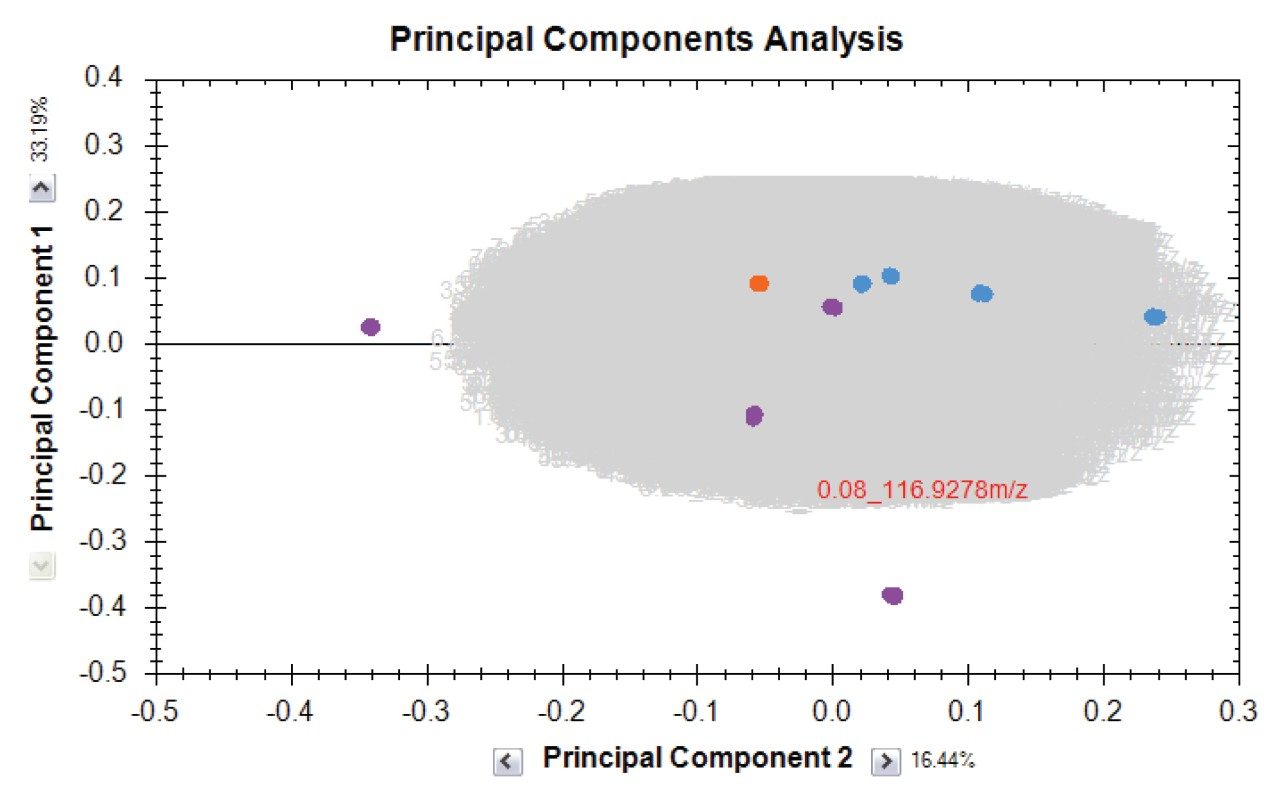
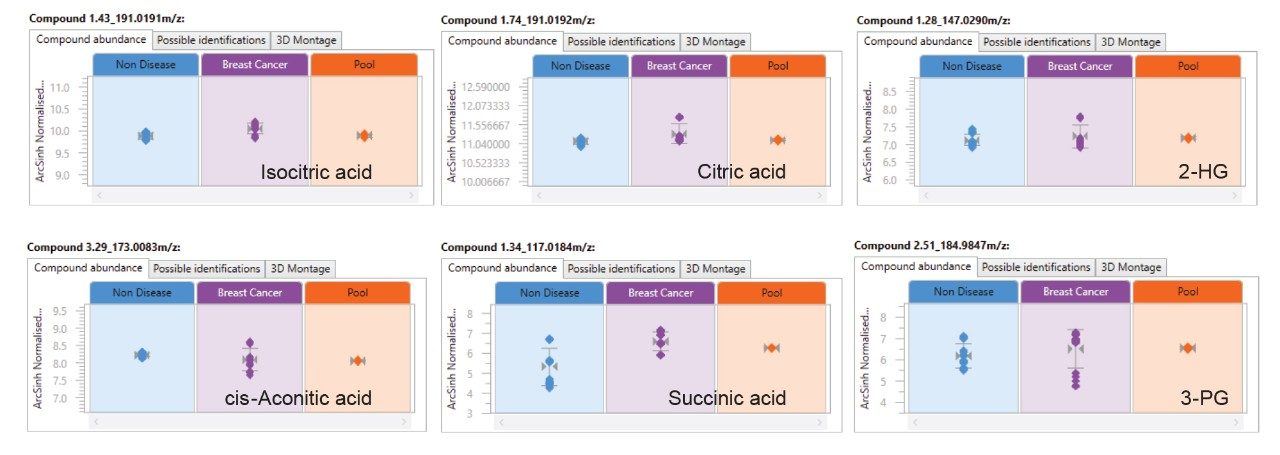
Here we present a mixed-mode LC method that is MS compatible for the analysis of TCA cycle analytes as well as other related compounds without the use of sample derivatization or ion-pairing reagents. The method incorporates the use of a column with hybrid organic inorganic surface technology, MaxPeak High Performance Surfaces (HPS), to mitigate analyte interactions with metal surfaces. We applied this method for the analysis of urine from healthy, and breast cancer subjects. We further used statistical software tools in order to determine any differences in analytes present as well as up- and down-regulated analytes in theses samples.