Automated High-Throughput LC-MS Focused Peptide Mapping of Monoclonal Antibodies in Microbioreactor Samples
Abstract
To facilitate cell line selection and bioprocess optimization, an automated high-throughput (HT) sample preparation method and fast LC-MS peptide mapping for Critical Quality Attribute (CQA) peptide measurement was developed for low volume and low concentration microbioreactor samples. For this method, 30 µL of a 1 mg/mL sample of a monoclonal antibody (mAb) in neutralized Protein A affinity chromatography elution buffer was buffer exchanged and trypsin digested using Andrew+™ automation. The resulting peptides were separated on an ACQUITY™ Premier Peptide CSH™ C18 Column and detected by a BioAccord™ LC-MS System. Effective analysis was demonstrated for selected CQA peptides including N-glycosylated, deamidated, and oxidized peptides. The automated preparation of 48 samples takes 3.5 hours, with a two-hour digestion time, and LC-MS analysis time was ten minutes per sample. Besides Protein A purified mAb, this method could also be adapted to the analysis of other samples with limited amounts of protein.
Benefits
- Consistent results for the relative quantification of CQA peptides from 30 µg of mAb even at low concentrations
- An automated trypsin digestion protocol with the Andrew+ robotic platform and using a new commercialized trypsin (RapiZyme™ Trypsin) that is highly resistant to autolysis
- High-throughput (HT) LC-MS analysis of CQA peptides using an ESI-ToF BioAccord LC-MS and automated data analysis using the waters_connect™ Peptide MAM application software
Introduction
Recombinant protein biopharmaceuticals, such as monoclonal antibodies (mAbs), have benefited patients for many years. Development and production of recombinant protein can be costly, and one potential bottleneck in developing an efficient process producing high quality product can be host cell-line selection and bioprocess optimization. To reduce the timeline for cell line selection and process optimization, microbioreactors have been used in early process development.1 In recent years, product quality has been included in cell line selection criteria, driven in part by the regulatory demands of making biosimilars. While microbioreactors provide advantages of being able to approximate the production conditions of a large-scale bioreactor, the amount of sample taken from a microbioreactor can be analytically limiting. Another challenge is that analyzing CQAs of the sample such as site-specific modifications usually involve peptide mapping via LC-UV or LC-MS, which can be labor-intensive and low throughput.
This application note demonstrates an automated procedure using an Andrew+ robot (Andrew Alliance™) with minimum manual intervention for a CQA peptide mapping method to assist cell line selection and cell-culture optimization (Figure 1). Presented is a focused or targeted peptide mapping method for monitoring the abundances of selected CQA peptides and their modified forms. For demonstration purposes, 30 µg of 1 mg/mL mAb (infliximab) sample was reduced and trypsin digested, after which its CQA peptides were monitored using a ten minute long reversed-phase separation with MS detection. This method could also be adapted for use with mAb and other protein samples with concentrations lower than 1 mg/mL, due to a pre-concentration step in the sample preparation procedure.
 Figure 1. Image of Andrew+ Pipetting Robot and BioAccord LC-MS System.
Figure 1. Image of Andrew+ Pipetting Robot and BioAccord LC-MS System.
Experimental
Sample Description
Infliximab (10 mg/mL) was diluted into neutralized Protein A elution buffer which contains 100 mM glycine (pH 3) and 1 M Tris (pH 7.5) in a 5:1 v/v ratio. The final infliximab concentration was 1.0 mg/mL.
For the stressed sample experiments, infliximab (10 mg/mL) was incubated in 0.005% H2O2 and 50 mM sodium phosphate (pH 7.6) for two weeks at 37 °C to induce oxidation and deamidation. The stressed sample was also co-mixed with original unstressed sample in 1:1 volume ratio. All samples were then diluted in the above neutralized Protein A elution buffer to 1.0 mg/mL.
LC Conditions
|
LC system: |
ACQUITY UPLC™ I-Class PLUS |
|
Detection: |
ACQUITY BioAccord MS System |
|
Plates: |
Acroprep™ Advance 350 µl Omega 10 k MWCO (p/n: PALL-8034) Eppendorf twin.tec® PCR Plate 96, skirted, 150 µL (p/n: 951020443) 6mm Pre-Slit Silicone/PTFE Cap Mat (Analytical Sales and services, p/n: 96727) |
|
Column(s): |
ACQUITY Premier Peptide CSH C18 1.7 µm, 2.1 x 100 mm (p/n: 186009488) |
|
Column temperature: |
60 ˚C |
|
Sample temperature: |
10 ˚C |
|
Injection volume: |
5 µL, 10 µL |
|
Flow rate: |
0.2 mL/min, 0.4 mL/min |
|
Mobile phase A: |
0.1% formic acid in water |
|
Mobile phase B: |
0.1% formic acid in acetonitrile |
Gradient Table (50-min gradient, 80-minute run time)
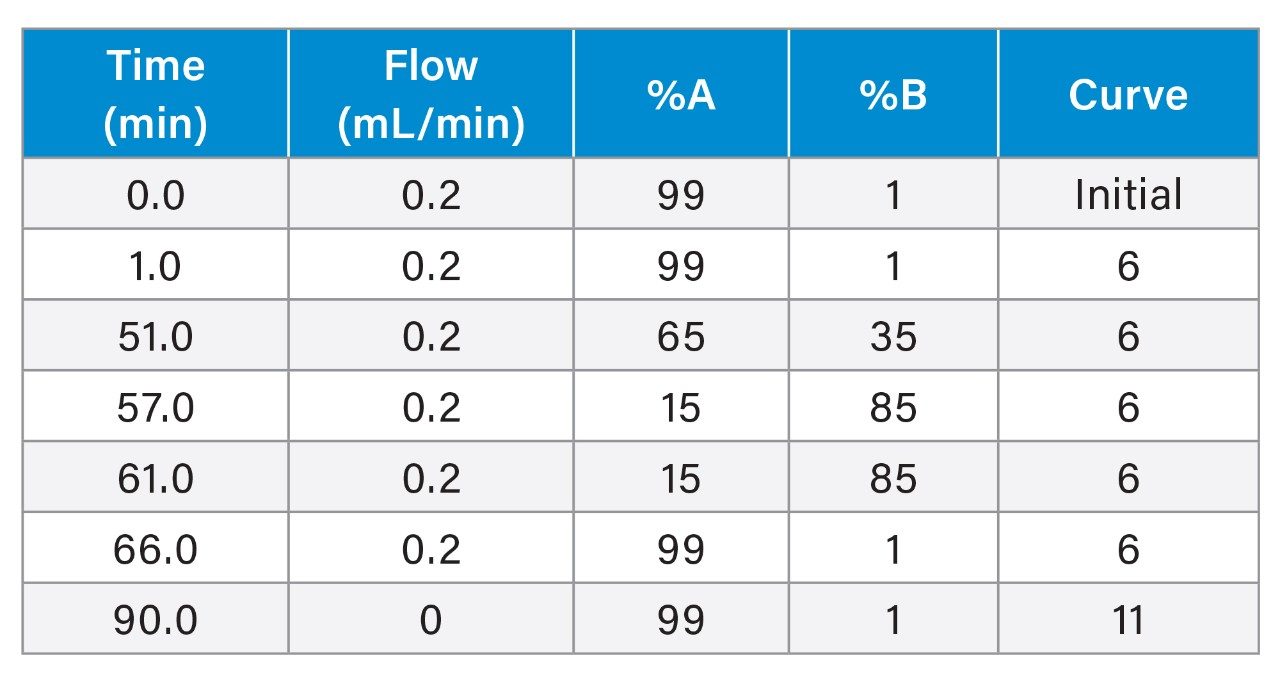
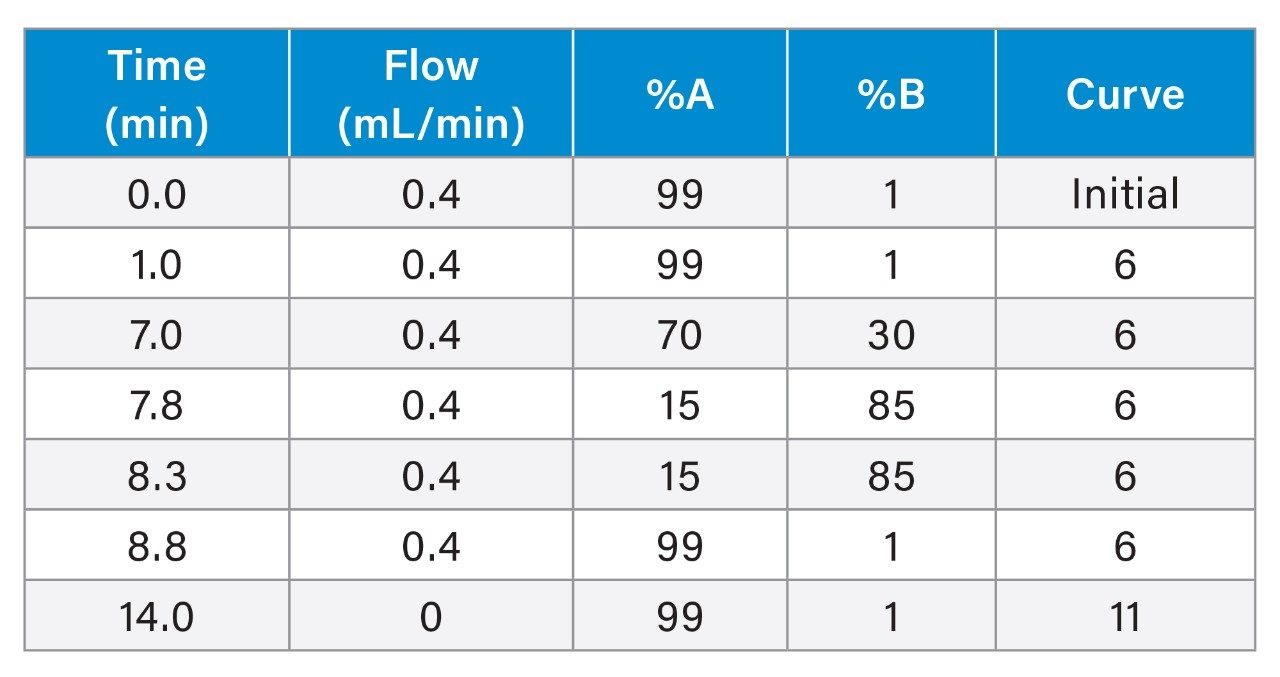
Gradient Table (6-min gradient, 10-minute run time)
ACQUITY RDa Detector Settings
|
Mode: |
Full scan with fragmentation |
|
Mass range: |
50–2000 m/z |
|
Polarity: |
Positive |
|
Sample rate: |
5 Hz |
|
Cone voltage: |
30 V |
|
Fragmentation cone voltage: |
60 V – 120 V |
|
Capillary voltage: |
1.20 kV |
|
Desolvation temperature: |
350 °C |
Data Management
|
LC-MS software: |
waters_connect |
Results and Discussion
General Procedure
An automated high throughput (HT) peptide mapping method using LC-MS analysis to monitor CQA peptides was successfully developed for low volume and low concentration microbioreactor mAb samples. Peptide mapping has been used for multiple attribute monitoring (MAM) by pharmaceutical industry for many years.2 It is typical to use 100 µg of protein or more in these digestion procedures because large amount of concentrated sample is often available for product characterization studies. However, in the current application, the samples that obtained from microbioreactors usually have low concentrations and volumes. To achieve an effective focused peptide mapping sample preparation, optimization of parameters including guanidine-HCl concentration, protein to enzyme ratio, digestion time was carried out (data not shown). It is important to note that the presented methods were not developed with the intent of comprehensively mapping the mAb, as such, significant under digestion is observed for these digest conditions. Also, the sample is only disulfide bond reduced without alkylation as part of this procedure.
Figure 2A shows the major steps for the automated focused peptide mapping procedure. Thirty µg of infliximab was diluted in neutralized Protein A elution buffer to 1.0 mg/mL (See EXPERIMENTAL for details). It is assumed that the mAb is affinity-purified with Protein A affinity chromatography resin.
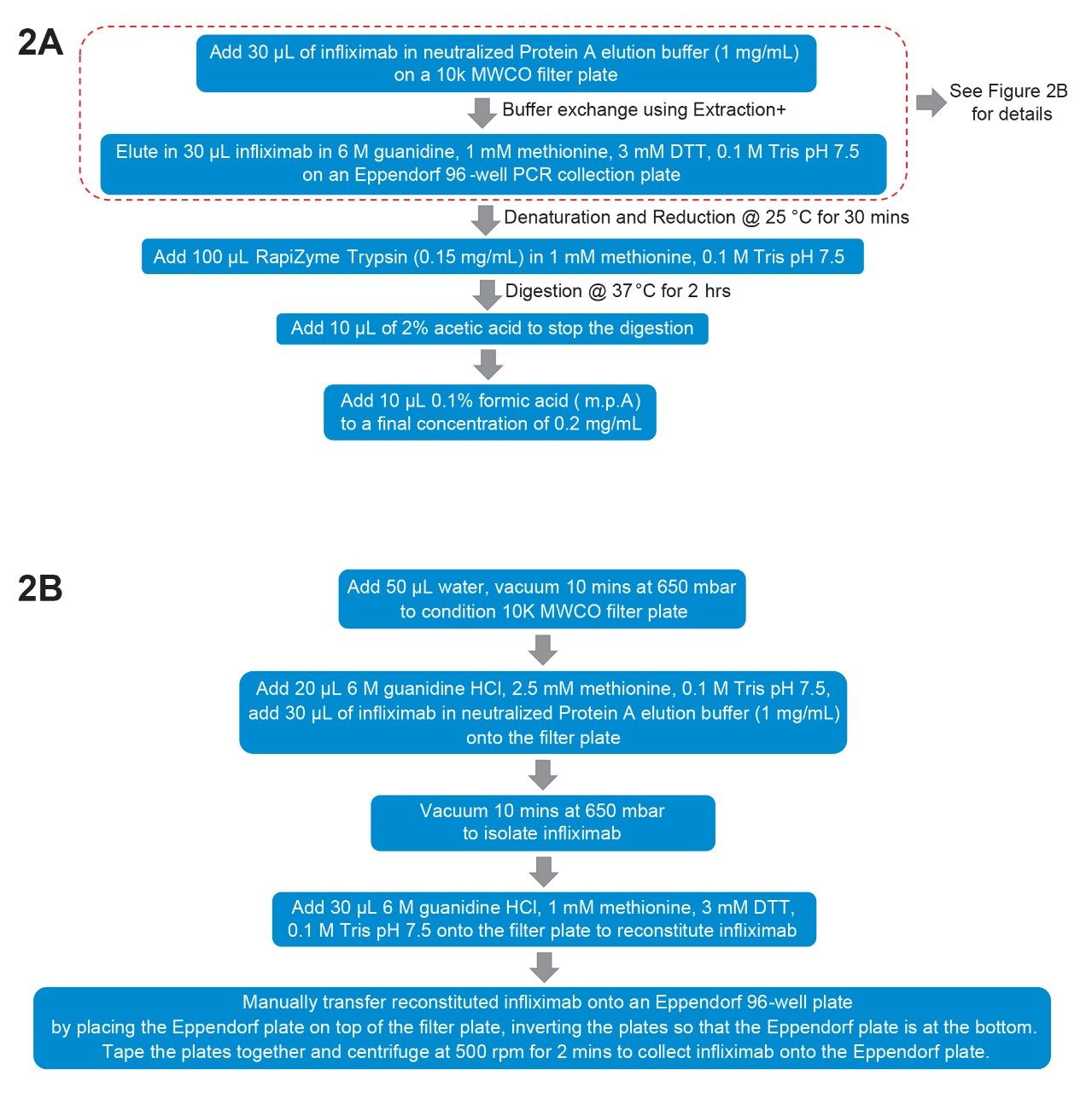 Figure 2. A. General digestion procedure using Andrew+ automation;
Figure 2. A. General digestion procedure using Andrew+ automation;B. Detailed procedure of buffer exchange.
For this method, the first step is to buffer exchange the samples into the denaturation and reduction buffer (DRB, 6 M guanidine HCl, 1 mM methionine, 3 mM DTT, and 0.1 M Tris pH 7.5) as shown in Figure 2B. This is done using Extraction+ domino on the Andrew+ automation system and a 10 K MWCO filter plate to retain the sample. Before the samples are loaded, 50 µL water is added to the filter plate wells and vacuum applied for ten minutes at 650 mbar to drain the water through the filter. It was discovered that this conditioning step can expedite the buffer drainage in the next steps.3 Then 20 µL of 6 M guanidine HCl, 2.5 mM methionine, 0.1 M Tris pH 7.5 solution and 30 µL of mAb (1.0 mg/mL) is added and vacuum is applied for 12 minutes at 650 mbar to drain the buffers while retaining the mAb. DRB (30 µL) is then added into the wells. To transfer samples from the filter plate to an Eppendorf 96-well PCR plate, the filter plate is inverted over the PCR collection plate and centrifuged at 500 RPM for two minutes. It is important to note that this inversion and centrifugation requires a user action and results in changing the well positions of the samples in a mirror image fashion.
After denaturation and reduction for 30 minutes at 25 °C, 100 µL 0.15 mg/mL RapiZyme Trypsin, a modified trypsin, in 1.0 mM methionine and 0.1 M Tris pH 7.5 is added to lower the guanidine-HCl concentration while digesting the mAb. The reaction mix is incubated at 37 °C for two hours, after which 10 µL of 2% acetic acid is added to stop the digestion. Finally, 0.10 % formic acid (mobile phase A) is added to dilute the reaction mix so that the final concentration of the digested protein is approximately 0.20 mg/mL.
A few points are worth noting for this procedure. First, the buffer exchange step can potentially pre-concentrate the sample if the sample concentration is low. In current experiments, for the purpose of demonstration, 30 µL sample was loaded and 30 µL was recovered. However, if the sample concentration is low, more volume can be loaded, and recovery can be done using less volume to concentrate the sample. Secondly, comparable results were obtained with and without alkylation. Therefore, alkylation step is not included in the procedure. Thirdly, the trypsin used in this procedure has an advantage of minimal low autolysis, so it can be used at high concentrations to speed up the digestion rate. Figure 3A and 3B shows the blank digest and the infliximab digest run on an ACQUITY Premier Peptide CSH C18, 2.1 x 100 mm Column with a 50-min gradient, respectively. Note: An ACQUITY Premier Peptide CSH C18 Column was selected to help ensure column to column performance consistency by QC testing each batch of synthesized CSH C18 particles with a tryptic protein digest rejecting those batches that do not meet performance specifications. The blank digest does not show many interfering trypsin peaks even though the Rapizyme Trypsin amount is high with a protein to enzyme ratio of 2:1.
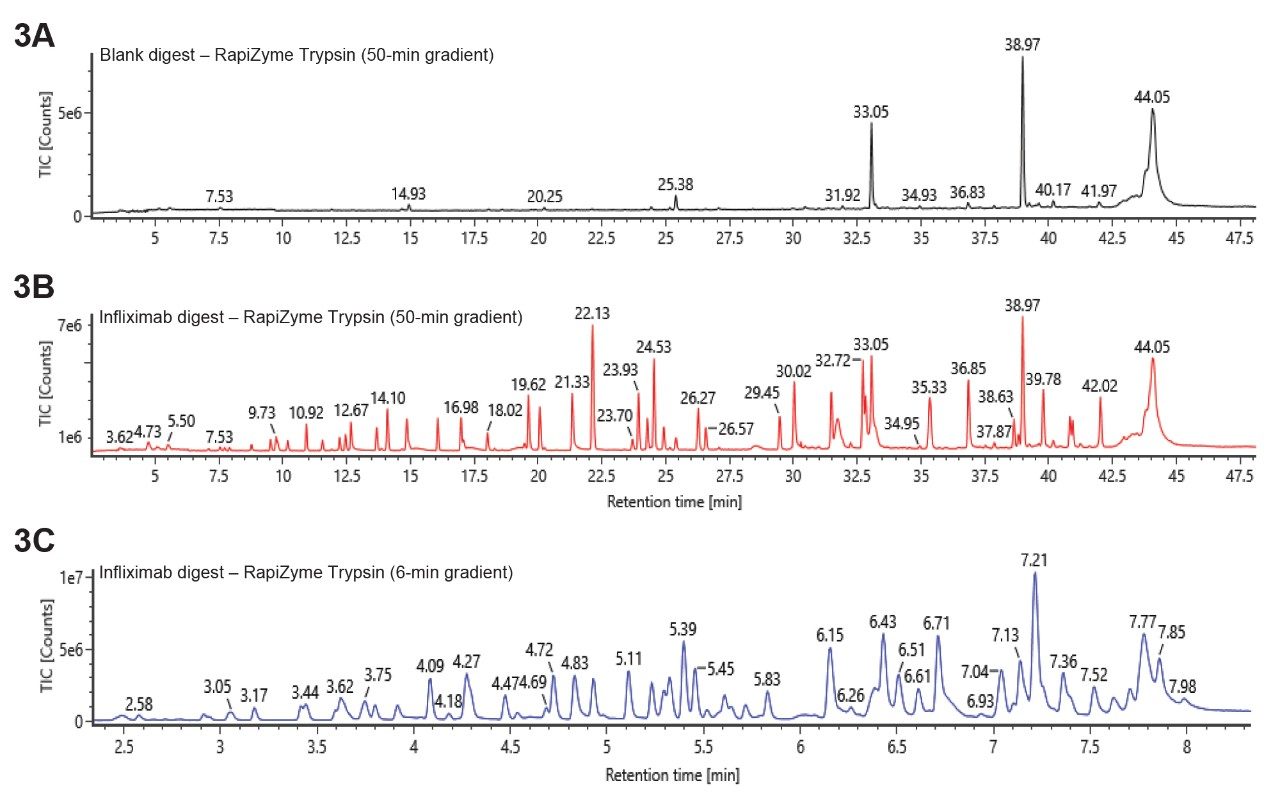 Figure 3. LC-MS chromatograms of infliximab CQA peptide mapping by RapiZyme Trypsin.
Figure 3. LC-MS chromatograms of infliximab CQA peptide mapping by RapiZyme Trypsin.A. Blank digest;
B. infliximab digest;
C. infliximab digest.
For A and B, the gradient is 1–35%B in 50 mins, 0.2 mL/min. For C, the gradient is 1–30%B in 6 mins, 0.4 mL/min.
For high throughput analysis, the digest was run with a 6-min elution gradient. As predicted, the 6-min gradient (Figure 3C) resulted in lower chromatographic resolution than the 50-min gradient (Figure 3B). Since the goal of this method is to enable relative quantitation of CQA peptides, calculated using combined ions counts from observed charge states, instead of fully characterizing the protein, it was found that the 6-min gradient was adequate.
Figure 4 shows the layout of Andrew+ for this experiment. It takes <3.5 hours to digest 48 samples using Andrew+ automation and eliminates seven manual pipetting steps.
 Figure 4. Andrew+ layout, dominos, and experimental time for digesting 48 samples.
Figure 4. Andrew+ layout, dominos, and experimental time for digesting 48 samples.
Reproducibility
The automated sample preparation and LC-MS analysis demonstrated acceptable reproducibility for a range of CQA peptides. For this study, samples of infliximab (30 µL of 1.0 mg/mL) were digested using a 48-sample protocol. Of these 10 samples representing different positions on the plate were chosen for LC-MS analysis (Figure 5). Automatic data analysis was executed using the waters_connect informatics platform and the Peptide MAM application software. Table 1 and Figure 6 show the relative abundances of the CQA peptides that were evaluated, and the relative standard deviations of those measurements. An injection volume of 10 µL (2 µg mAb) was found to result in more consistent results than injecting 5 µL (1 µg). In this example, injecting more than 2 µg did not improve the reproducibility results further. For all 20 CQA peptides, %RSD of the percent modification was < 20% (n=10), and of the eight CQA peptides with a %RSD between 10% to 20%, seven were at levels of 1.66% or lower. The cause of slightly higher RSD% for oxidated peptides is under investigation. On the whole, these data demonstrate that this HT LC-MS method can provide consistent results of relative abundance of site-specific modifications of a protein using limited amounts (30 µg) sample.
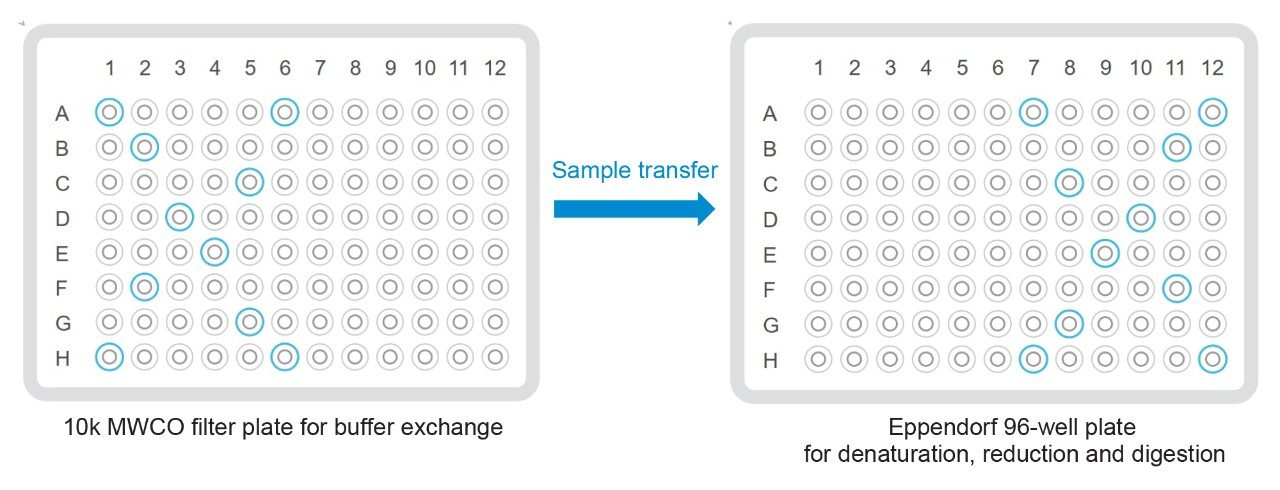 Figure 5. Positions of 10 representative wells for reproducibility study. Notice that the positions of the wells are mirror-images before and after buffer exchange.
Figure 5. Positions of 10 representative wells for reproducibility study. Notice that the positions of the wells are mirror-images before and after buffer exchange.
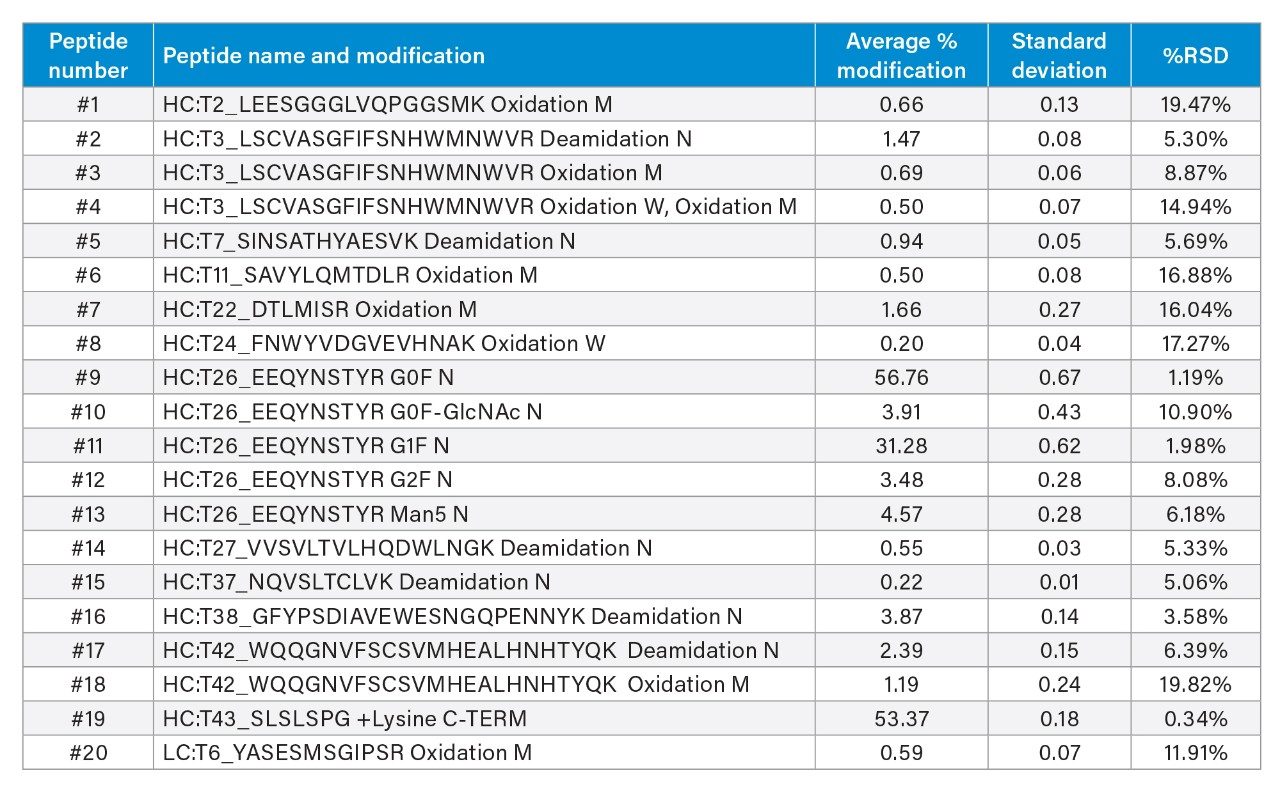 Table 1. Average, standard deviation, and %RSD of relative abundances of infliximab CQA peptides (n=10).
Table 1. Average, standard deviation, and %RSD of relative abundances of infliximab CQA peptides (n=10).
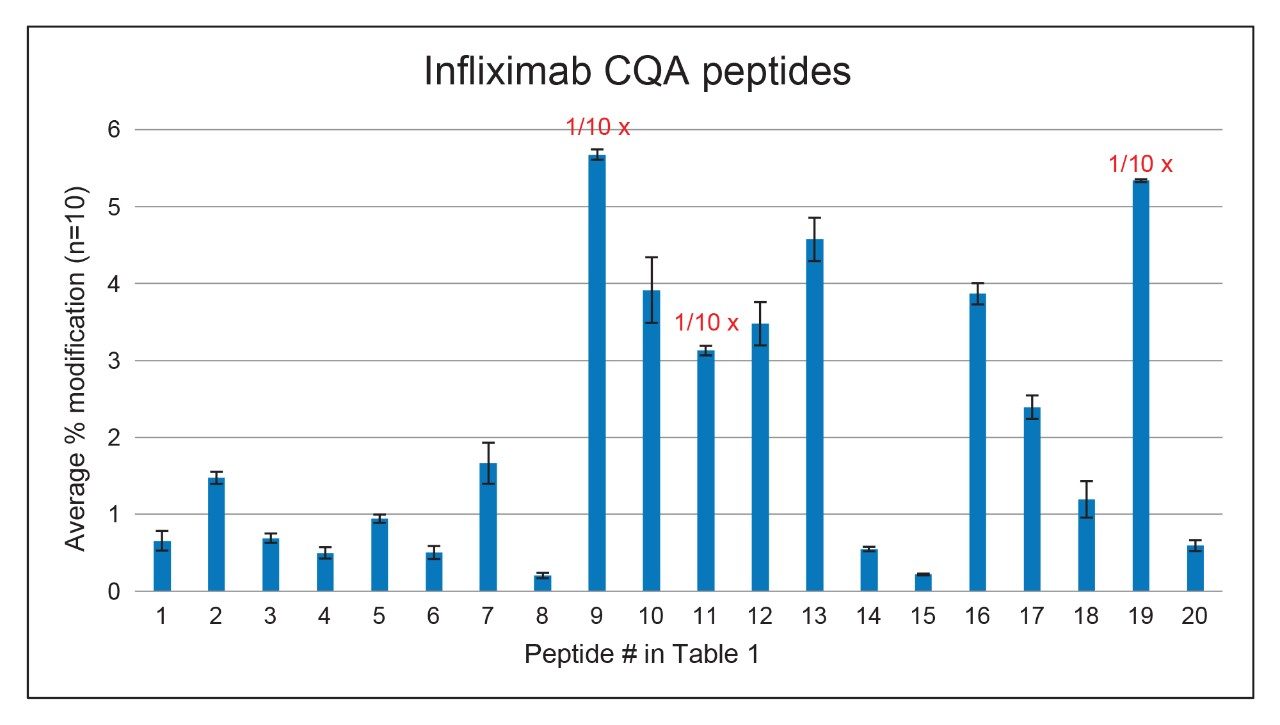 Figure 6. Average and standard deviation of relative abundances of infliximab CQA peptides (n=10). For peptide #9, #11 and #19, one tenth of the signal was plotted.
Figure 6. Average and standard deviation of relative abundances of infliximab CQA peptides (n=10). For peptide #9, #11 and #19, one tenth of the signal was plotted.
Stressed Sample Analysis
For cell line selection and process optimization, it is important that the method can detect changes of the percent modification of CQA peptides. For a fit for purpose demonstration, samples were stressed to increase oxidation and deamidation (please see EXPERIMENTAL for details). Stressed sample was also mixed with un-stressed sample (1:1 volume ratio). Figure 7A shows percent modification of several infliximab CQA peptides. The LC-MS data for the four oxidized and three deamidated CQA peptides evaluated were consistent with the 1:1 co-mixed sample having intermediate levels of degraded peptides. The sensitivity of this analysis to detect CQA changes is best exemplified in the results observed for the deamidated HC:T37 peptide NQVSLTCLVK (f in Figure 7A), which increased in abundance from 0.1% to 0.6%. The mass spectra of un-modified (top) and deamidated (bottom) form of this peptide are shown in Figure 7B. Based on the retention time, this deamidated form is likely aspartic acid that is converted from asparagine. Figure 7C shows results for the N-glycan modified and C-term heavy chain (HC) peptides. The HC C-terminal peptide can be present with or without a lysine residue at the C-terminus. As predicted, consistent percent modification was obtained for these stable peptide modifications. Overall, the results show that this automated HT CQA peptide mapping method is able to detect site-specific changes among different bioprocessing samples.
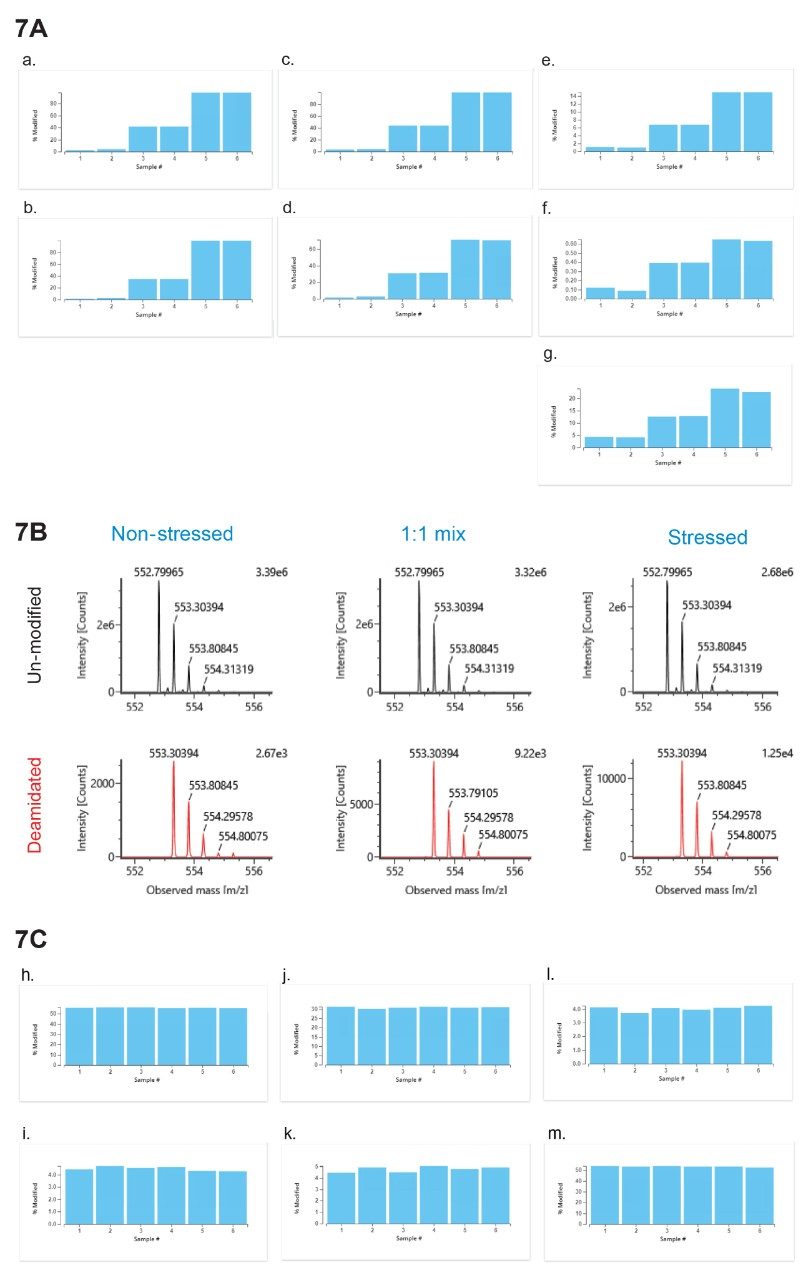 Figure 7. Percent modification of infliximab CQA peptides. #1 and #2 are un-stressed samples, #5 and #6 are stressed samples, while #3 and #4 are the 1:1 mixed samples.
Figure 7. Percent modification of infliximab CQA peptides. #1 and #2 are un-stressed samples, #5 and #6 are stressed samples, while #3 and #4 are the 1:1 mixed samples. A. For CQA peptides that have oxidation or deamidation modification, the percent modification of the un-stressed and stressed sample 1:1 volume mix is approximately in the middle of the percent modification of the un-stressed sample and the stressed sample. a. HC:T2 (LEES…GSMK) Oxidation M; b. LC:T6 (YASE…IPSR) Oxidation M; c. HC:T22 (DTLMISR) Oxidation M; d. HC:T42 (WQQG…TYQK) Oxidation M; e. HC:T7 (SINS…ESVK) Deamidation N; f. HC:T37 (NQVS…CLVK) Deamidation N; g. HC:T38 (GFYP…NNYK) Deamidation N.
B. Mass spectra of HC:T37 peptide NQVSLTCLVK (f in Figure 7A). Top: un-modified form (m/z: 552.81, doubly charged); Bottom: deamidated form (m/z: 553.30, doubly charged).
C. For CQA peptides that do not have oxidation or deamidation modification, the percent modification is consistent regardless of the stress state. h: HC:T26 (EEQYNSTYR) G0F N; i: HC:T26 (EEQYNSTYR) G0F-GlcNAc N; j: HC:T26 (EEQYNSTYR) G1F N; k: HC:T26 (EEQYNSTYR) Man5 N; l: HC:T26 (EEQYNSTYR) G2F N; m. HC:T43 (SLSLSPG) +Lysine C-TERM.
Conclusion
Consistent results for the relative quantification of CQA peptides using 30 µg of mAb sample were obtained using a method featuring an automated trypsin digestion protocol with the Andrew+ robotic platform and using a new trypsin, RapiZyme Trypsin, that is highly resistant to autolysis, and offers increased activity compared to other sequencing grade trypsin products.4,5 This automated trypsin digest method is capable of generating 48 samples within four hours. In addition, the ESI-ToF BioAccord LC-MS method has a ten minutes total run time and automated LC-MS data processing is delivered by waters_connect using the Peptide MAM application software.
The general procedures outlined in this application note demonstrate the capabilities of the robotic platform and LC-MS used but can be readily optimized to meet the specific analytical requirements of other protein samples.
References
- Lange I., Chhatre S., and Zoro B. Reducing Timelines in Early Process Development – Using a Multiparametric Clone-Selection and Feed-Optimization Strategy. Bioprocess International, November 2014.
- Ren D., Pipes G.D., Liu D., Shih L-Y, Nichols A.C., Treuheit M.J., Brems D.N., Bondarenko P.V. An improved trypsin digestion method minimizes digestion-inducedmodifications on proteins. Analytical Biochemistry 392 (2009) 12–21.
- Hanna C. M., Koza S. M., Yu Y. Q. Automated High-Throughput N-glycan Labelling and LC-MS Analysis for Protein A Purified Monoclonal Antibodies. Waters Application note. 720007854, February 2023.
- Ippoliti S., Zampa N., Yu Y. Q., Lauber M. A. Versatile and Rapid Digestion Protocols for Biopharmaceutical Characterization Using RapiZyme™ Trypsin. Waters Application note. 720007840, January 2023.
- Finny A. S., Zampa N., Addepalli B., Lauber M. A. Fast and Robust LC-UV-MS Based Peptide Mapping Using RapiZyme™ Trypsin and IonHance™ DFA. Waters Application note. 720007864, February 2023.
720007885, April 2023